
Synthesis and evaluation of novel non-covalent binding quinazoline glycoside derivatives targeting the L858R and T790M variants of EGFR†
Renshuai Zhanga,
Shaopeng Chena,
Xiaowei Zhangb,
Rilei Yua,
Shengbiao Wana,
Meiyu Geng*b and
Tao Jiang*a
aKey Laboratory of Marine Drugs, The Ministry of Education of China, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China. E-mail: jiangtao@ouc.edu.cn
bDivision of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, P. R. China. E-mail: mygeng@simm.ac.cn
First published on 7th April 2016
Abstract
A series of novel quinazoline glycoside derivatives were designed, synthesized, and evaluated for their inhibition activities against EGFR-WT, EGFR/L858R/T790M, and skin epidermoid carcinoma cell line (A431). Several L-rhamnose derivatives were found to be as efficient as the irreversible inhibitor HKI-272 or BIBW2992 in inhibiting EGFR/T790M/L858R. Molecular dynamic simulations indicated that the saccharide group plays a significant role in stabilization of the quinazoline glycoside derivative through the hydrogen bonding with several polar residues at the edge of the ATP-binding cleft.
It is well established that receptor protein tyrosine kinases (PTKs) play a crucial role in activating numerous signal transduction pathways within cells, leading to cellular proliferation, differentiation and various regulatory mechanisms.1,2 EGFR is a member of the ErbB family of PTKs.3 Overexpression of EGFR is associated with poor prognosis in many human cancers.4,5 Thus, EGFR tyrosine kinase is an attractive therapeutic target for cancer treatment.
The first-generation of EGFR inhibitors, gefitinib (Fig. 1), have demonstrated significant clinical benefit for non-small cell lung cancer (NSCLC) patients harboring activating mutations in EGFR through the reversible inhibition of EGFR tyrosine kinases.6–8 Nevertheless, the clinical efficacy of them is limited by the development of drug-resistance mutations, including the gatekeeper T790M mutation, which accounts for approximately 50% of the population of the clinically acquired resistant patients.9,10 The second-generation irreversible EGFR inhibitors (HKI-272 and BIBW2992 in Fig. 1) and the third-generation irreversible inhibitors (AZD9291) could overcome the acquired resistance caused by the T790M mutation.10–12 However, these irreversible EGFR inhibitors are still discouraging due to their limited clinical efficacy associated with skin rash and gastrointestinal toxicity.13,14
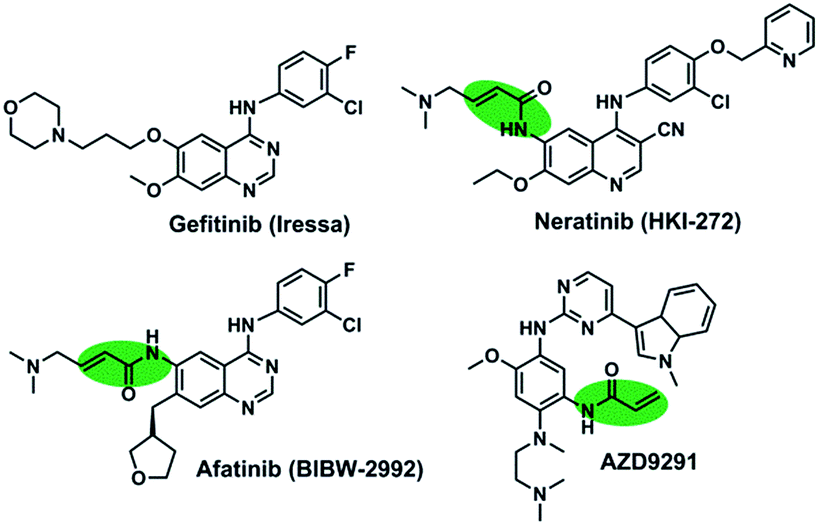 | ||
| Fig. 1 Chemical structures of reversible and irreversible EGFR inhibitors. | ||
These irreversible inhibitors contain an acrylamide functionality that forms a covalent bond with Cys797.15,16 The covalent bond is attributed to the improved activity of these compounds in cellular models with T790M mutant. However, at least ten kinases, including EGFR, have a reactive cysteine residue in the position equivalent to Cys-797 in EGFR, and thus they are potential targets of irreversible inhibitors containing acrylamide fragment. Thus, the potential toxicity of the covalent inhibitors caused by off-target effects should be concerned. Moreover, many studies indicated that the covalent bond between Cys-797 and acrylamide fragment is not required for effective inhibition of the T790M mutant. It is, therefore, possible that a reversible inhibitor should work as well if it binds with sufficient affinity to out-compete ATP.17
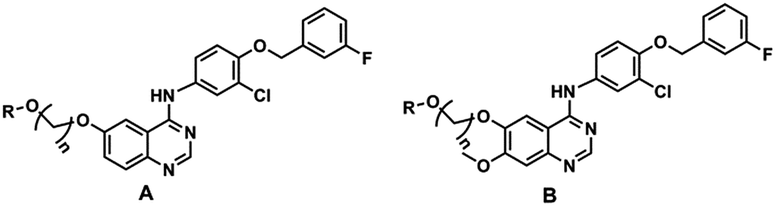
In this paper, we present a series of novel T790M mutant kinase reversible inhibitors without an acrylamide functionality that presents in HIK272 and BIBW-2992. The designed inhibitors compose of three parts: anilinoquinazoline core, saccharide moiety and a suitable linker between quinazoline core and saccharide moiety (Fig. 2).
 | ||
| Fig. 2 Design of a series of quinazoline glycoside derivatives. | ||
The key intermediate 10a was synthesized from 2-nitro-5-methoxybenzoic acid (see ESI Scheme S1†). The glycosyl donor 10 was synthesized by coupling full acetylated glucose with the 2-bromoethanol, as described in ESI Scheme S2.† Compounds 20 to 39 were synthesized as shown in Scheme 1. The glycosylation of compound 10a using compound 10 as glycosyl donor in the presence of K2CO3 at 80 °C provided compound 20a, which was deacetylated by sodium methoxide in methanol to yield target compound 20 successfully.17–22 The compounds 21 to 39 were prepared by a similar method to that described for compound 20 synthesis.
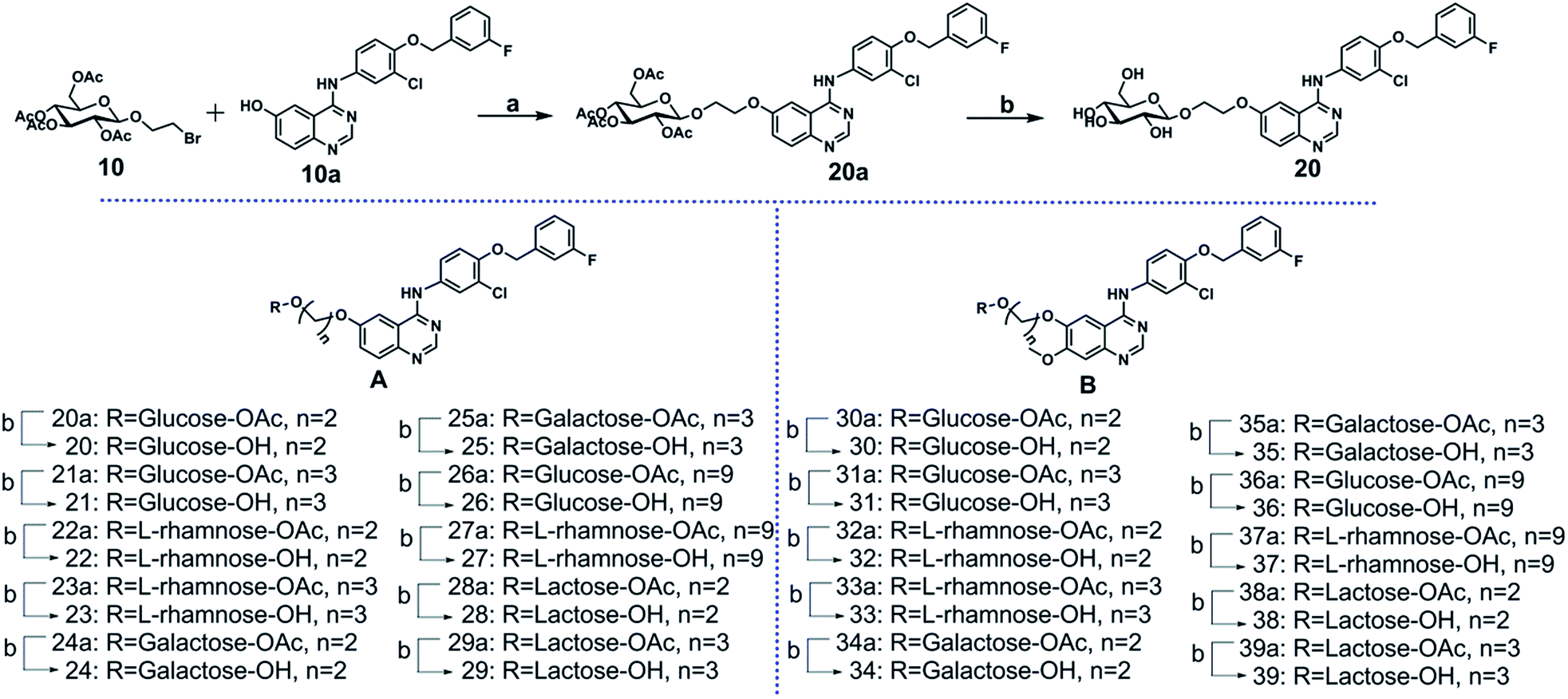 | ||
| Scheme 1 Reagents and conditions: (a) K2CO3, anhydrous DMF/80 °C, 7 h; (b) NaOMe/MeOH, rt. | ||
The inhibition activity of the designed compounds against EGFR and A431 was evaluated (Table 1). Encouragingly, most of the glycosylated derivatives potently suppressed the EGFR-WT kinases activity with potency comparable to that of gefitinib. In addition, more than half of the compounds displayed significant potency in the anti-proliferation assay of A431 cell line. The SAR of glycoside derivatives was summarized. First, we investigated the effect of the linker on the activity of A-series compounds against EGFR-WT. When the R moiety was glucose, the best linker was three carbon alkyl chains (n = 3). Under the experimental conditions, the compound 21, a substituted derivative of glucose with three carbon atoms linker chain, potently inhibited EGFR-WT with IC50 values of 1.65 nM, which were more potent than those of compounds 20 (n = 2) and 26 (n = 9). The same variation was also observed when the R moiety was L-rhamnose. The compounds (20–23) containing two or three carbon alkyl chains showed similar activity against EGFR, however they were more potent than derivatives (26, 27) containing nine carbon alkyl chains. B-series compounds 30–37, contained 3-chloro-4-(3′-fluorobenzyloxy) aniline and 7-methoxy substituents, were only slightly more potent in inhibiting EGFR-WT compared to the parent A-series of quinazoline derivatives. Consistent with the result that observed in A series of compounds, compounds 30–33 with linker of 2–3 carbon displayed more potent than compounds 36 and 37, which containing a linker of nine carbon. Next, the effect of the glycosylation moiety on the activity was also investigated. It was found that the glucose, L-rhamnose, galactose and lactose derivatives were almost equally potent against EGFR-WT in A and B series of compounds.
|
|||||
|---|---|---|---|---|---|
| Cmpd | Series | R | n | EGFR-wt IC50 (nM) | A431 IC50 (μM) |
| a NA: no activity (IC50 > 50 μM). | |||||
| 20 | A1 | Glucose | 2 | 3.64 ± 0.12 | 7.58 ± 1.26 |
| 21 | A2 | Glucose | 3 | 1.65 ± 0.16 | 6.61 ± 2.15 |
| 22 | A3 | L-Rhamnose | 2 | 5.28 ± 0.78 | 2.69 ± 0.34 |
| 23 | A4 | L-Rhamnose | 3 | 3.94 ± 0.64 | 2.15 ± 0.22 |
| 24 | A5 | Galactose | 2 | 3.16 ± 0.82 | NAa |
| 25 | A6 | Galactose | 3 | 5.29 ± 1.01 | NA |
| 26 | A7 | Glucose | 9 | 7.50 ± 2.10 | 39.81 ± 0.57 |
| 27 | A8 | L-Rhamnose | 9 | 14.00 ± 1.25 | 26.19 ± 0.82 |
| 30 | B1 | Glucose | 2 | 5.29 ± 1.09 | NA |
| 31 | B2 | Glucose | 3 | 0.54 ± 0.18 | NA |
| 32 | B3 | L-Rhamnose | 2 | 0.70 ± 0.18 | 2.75 ± 1.37 |
| 33 | B4 | L-Rhamnose | 3 | 0.84 ± 0.14 | 2.59 ± 0.88 |
| 34 | B5 | Galactose | 2 | 0.14 ± 0.09 | NA |
| 35 | B6 | Galactose | 3 | 0.49 ± 0.07 | NA |
| 36 | B7 | Glucose | 9 | 12.60 ± 3.36 | 12.52 ± 4.19 |
| 37 | B8 | L-Rhamnose | 9 | 13.10 ± 0.33 | 6.94 ± 1.53 |
| Gefitinib | 39.00 ± 3.11 | 3.3 ± 0.12 | |||

Subsequently, the activity of compounds against A431 was analysized. The potency of L-rhamnose derivatives against A431 cell line was significantly more potent than those of glucose or galactose derivatives. For example, the compounds 21, 23 and 25 with the same linker (n = 3) but having different glycosylation moiety displayed entirely different potency against A431. The compound 23, an L-rhamnose derivative, exhibited the best activity against A431 cell, while the potency of compound 21 against A431 cell was considerably reduced, and the potency of compound 25 was almost entirely abolished. Similarly, the galactose compound 24 did not exhibit any activity on A431. Based on these results, we concluded that galactose was not a proper glycosylation moiety for improving activity. This may be due to that the quinazoline derivatives containing galactose could not be transported by cells. In B-series compounds, the inhibitory potency of different glycosyl derivatives against A431 cell line was not significantly changed compared to the corresponding A-series derivatives. The compounds 32 and 33 were the most potent inhibitors against A431 cell line, which were L-rhamnose derivatives with the linker of two or three carbon alkyl chain. Interestingly, this result was similar to that of A-series compounds.
Further structure modification was conducted to explore the role of saccharide. When the number of sugar ring increased in quinazoline compounds, more hydroxyl groups were introduced into the modified inhibitor, which possessed more potential donor or acceptor of the hydrogen bond. In principle, more hydroxyl groups could enhance the activity of new compounds. The compound 28, 29, 38 and 39 which were lactose derivatives, displayed excellent inhibitory activity against EGFR-WT with IC50 values in the low nanomolar ranges, which were better than gefitinib (Table 2). However, these compounds were completely inactive against cell line A431, which may be attributed to the reduced cellular permeability of lactose derivatives. Taking into account that activity was not enhanced by the modification of lactose, which introduced more donor and acceptor of the hydrogen bond, compounds 40 to 43, the hydrogen bond donor was reduced through protected part of the hydroxyl, were prepared as depicted in Scheme 2. The selective protection of 4,6-dihydroxyl in the glucose ring was achieved by treatment of compounds 20 with 2-ethoxypropene in the presence of catalytic amount of TsOH (p-toluenesulfonic acid) in anhydrous DMF, affording the target compound 40. Compound 41 to 43 were also synthesized in a similar procedure.22,23
|
|||||
|---|---|---|---|---|---|
| Cmpd | Series | R | n | EGFR-wt IC50 (nM) | A431 IC50 (μM) |
| a NA: no activity (IC50 > 50 μM). | |||||
| 28 | A9 | Lactose | 2 | 11.21 ± 0.77 | NAa |
| 29 | A10 | Lactose | 3 | 3.10 ± 1.62 | NA |
| 38 | B9 | Lactose | 2 | 0.84 ± 0.11 | NA |
| 39 | B10 | Lactose | 3 | 16.33 ± 0.45 | NA |
| 40 | A11 | PG-glucose | 2 | 4.9 ± 0.98 | 25.13 ± 2.17 |
| 41 | A12 | PG-glucose | 9 | 43.2 ± 2.69 | 29.24 ± 1.53 |
| 42 | B11 | PG-glucose | 2 | 1.50 ± 0.26 | NA |
| 43 | B12 | PG-glucose | 9 | 30.00 ± 0.76 | NA |
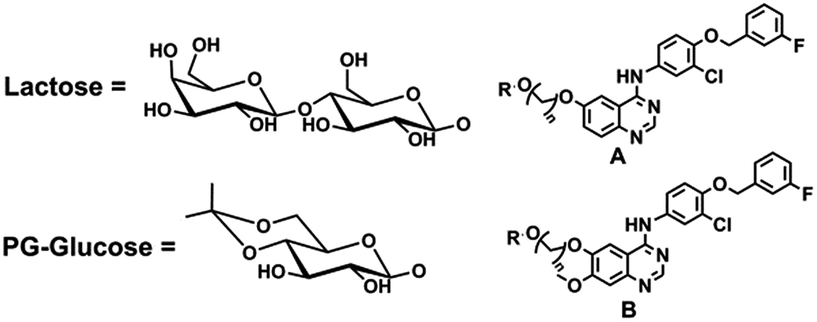
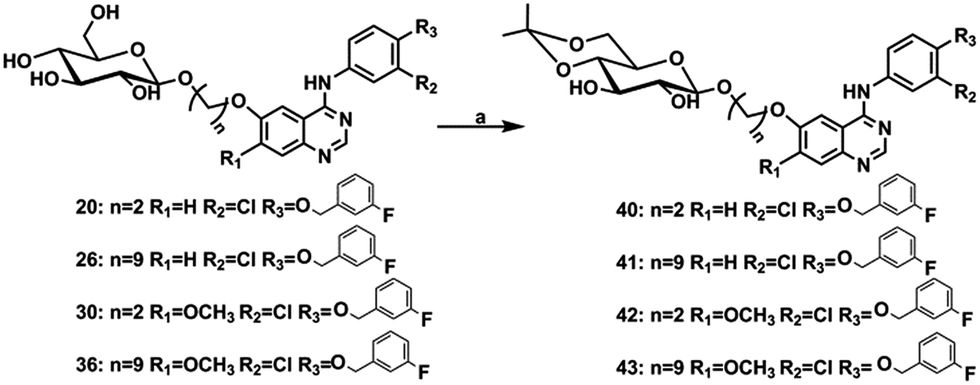 | ||
| Scheme 2 Reagents and conditions: (a) 2-ethoxypropene, TsOH·H2O, anhydrous DMF, calcium sulfate anhydrous, 30 °C, 2 h. | ||
The activity of compounds 40–43 was displayed in Table 2. The compounds 40 and 41, the 4 6-dihydroxyl of glucose ring was selectively protected, only exhibited a moderate activity on enzyme and A431 cell line, while compounds 42 and 43 were no activity against A431. These results suggested that this modification did not significantly improve the potency. In contrast, it reduced the suppressive function of modified compounds against A431.
Based on the result displayed in Table 1, the chemically modified of glucose or L-rhamnose and 2–3 carbon chain was favorable to activity. On the contrary, the galactose and lactose derivatives displayed disappointing results. Similarly, the linker of nine carbon atoms is not conducive to enhance activity. The effect of 7-methoxy substituents was ambiguous. However, the compounds 20 and 21 displayed different activity compared with 30 and 31, that was impressively. It is worth further study why methoxy affected only the activity of glucose derivatives. The results of Table 2 showed that the expansion of sugar ring, including the increase or decrease of hydrogen bond donors, was not conducive to improve activity. It perhaps affect the ability of compounds to penetrate cells.
To investigate the effect of saccharide ring, linker and 7-methoxy, molecular docking was adopted to predict the binding mode of the representative compound 23, 27 and 33. The docking simulations were carried out by means of the SYBYL-X 2.0 software, using the publicly available coordinate of the EGFR (PDB code: 4i23).
The binding modes of compounds 23, 27 and 33 were respectively displayed in (A), (B) and (C) of Fig. 3, where some main interactions between the ligand and the receptor can be observed. The overlay poses of compounds 23 and 33 was showed in Fig. 3D. It can be seen that the 4-anilino groups of compound 23, a substituted derivative of L-rhamnose with three carbon atoms linker, embedded in the hydrophobic pocket compromised of Val726, Ala743, Leu718 and Leu792 etc. This is consistent with that the binding mode of 4-anilino quinazoline compounds with EGFR. Moreover, the expected hydrogen bonds are formed between the saccharide moiety and several amino acids in the EGFR solvent region. The hydroxyl of L-rhamnose form hydrogen bonds with Lys750 and Asp855. The compound 27, L-rhamnose derivative containing nine carbon atoms linker, showed disappointing binding mode. The saccharide moiety of 27 form hydrogen bonds with Arg841, however there was no interaction between 4-anilino group and the hydrophobic pocket. That may explain why the activity against EGFR of 27 was less than 23. The Fig. 3C and D showed that the 4-anilino groups and 6-substituents of compounds 23 and 33 adopt a very similar conformation at the EGFR ATP binding pocket. The methoxyl make some impact on positioning of quinazoline core of 33 (Fig. 3D), and such that, 33 form more hydrogen bonds with EGFR (Fig. 3C).
 | ||
| Fig. 3 (A), (B) and (C) Low-energy docking mode of the WT-EGFR/23 (A), 27 (B), 33 (C) complex. The EGFR kinase is shown in a cartoon representation (green) with 23, 27 and 33 in stick model representation (gray white). H-bond interactions are shown as black dotted lines. (D) Overlay poses between 23 (stick model with carbons colored in green) and 33 (stick model with carbons colored in gray white). The ATP-binding site of the EGFR is shown in the transparent molecular surface colored by atom type representation. | ||
The toxicity profiles (including mutagenic effect, tumorigenic effect, irritating effect and reproductive effect) of compound 33, gefitinib and HKI-272 were predicted using OSIRIS Property Explorer software at URL http://www.organic-chemistry.org/prog/peo/. The prediction of toxicity risks were shown in Table 3. The glycoside derivative 33 and gefitinib showed low risk in toxicity prediction. In contrast, HKI-272 (irreversible inhibitors) showed high mutagenicity. That may be related to the acrylamide group, which could form a covalent bond with cysteine in some enzymes.
To assess further their potency, several selected compounds were tested against EGFR/L858R/T790M (Table 4). As expected, the compounds 23 and 33, L-rhamnose derivatives with a linker of three carbon alkyl chain, strongly inhibited EGFR/L858R/T790M at 1 μM concentration. The compound 23 showed similar activity compared with irreversible inhibitor HKI-272 and the compound 33 exhibited more potent than HKI-272 and BIBW-2992 (irreversible inhibitors).
|
|||||
|---|---|---|---|---|---|
| Cmpd | Series | R | n | EGFR/T790M/L858R% inhibition | |
| 10 μM | 1 μM | ||||
| a NT: no test. | |||||
| 20 | A1 | Glucose | 2 | 69.9 | 14.0 |
| 21 | A2 | Glucose | 3 | 89.6 | 42.7 |
| 22 | A3 | L-Rhamnose | 2 | 94.0 | 52.8 |
| 23 | A4 | L-Rhamnose | 3 | 93.5 | 77.9 |
| 40 | A11 | PG-glucose | 2 | 47.0 | NTa |
| 27 | A8 | L-Rhamnose | 9 | 9.1 | NT |
| 32 | B3 | L-Rhamnose | 2 | 92.7 | 42.3 |
| 33 | B4 | L-Rhamnose | 3 | 100 | 94.8 |
| 36 | B7 | Glucose | 9 | 49.2 | NT |
| 37 | B8 | L-Rhamnose | 9 | 23.1 | NT |
| HKI-272 | NT | 77.8 | |||
| BIBW2992 | NT | 80.7 | |||
To elucidate the mechanism of saccharide ring that contributes to the increased affinities of the quinazoline glycoside derivatives with EGFR, molecular docking and molecular dynamics (MD) simulations were performed to predict the binding mode of the representative potent compound 33. Interestingly, compound 33 was positioned at the EGFR ATP binding site in an orientation quite similar to those of HKI and lapatinib, in which the 4-aniline group deeply embedded in the hydrophobic pocket compromised of L718, V726, A743, M790, L777, M766, L788, and L747. On the other hand, the saccharide moiety suitably positioned at the gate of the pocket where located M793, F795, C797, and D800 (Fig. 4 and S1†). In addition, the H-bond between the N1 atom of the quinazoline and the HN atom on the backbone of M793 is also conserved among these three EGFR (mutant) complexes. The saccharide moiety forms H-bonds with backbone atoms of C797 and F795 (Fig. 4) as well as a less stable H-bond with D800 (not shown). These H-bonds significantly contribute to the high affinity of the quinazoline glycoside derivatives, consistent with experimentally tested inhibitory activity against the EGFR mutant. According to the results of the MD of 33, these H-bonds are stable in most of the trajectory, despite moderate fluctuation.
 | ||
| Fig. 4 The binding mode of compound 33 with EGFR/T790M/L858R. In panel (A), compound 33 was shown in orange, and the H-bonds of were shown in dashed lines. Panel (B), the distances between the H-bond acceptor and H-bond donor. | ||
Conclusions
In the present work, a series of novel quinazoline glycoside derivatives were designed, synthesized and evaluated. Some compounds showed satisfactory results in inhibiting EGFR-WT and were proved efficient in A431 lung cancer cell. We summarized the structure–activity relationship of the new quinazoline glycoside derivatives. In conjunction with the binding mode and MD, these results suggested that the best glycosylation was L-rhamnose and best linker was also 2 or 3 carbon atoms. The data that we have obtained revealed that the saccharide moiety and the length of the linker were the key factors in determining the activity of quinazoline glycoside derivatives. Notably, the potency of several L-rhamnose derivatives to suppress EGFR/T790M/L858R was similar to the irreversible inhibitors HKI-272 and BIBW-2992. Two L-rhamnose derivatives, without acrylamide functionality that could form covalent bonds, displayed potent activity against drug-resistance mutant EGFR. The compound 33 exhibited more potent than HKI-272 and BIBW-2992 (irreversible inhibitors). This is a preliminary proof that the glycosylation is a feasible strategy to design potent EGFR inhibitors. Future studies will focus on the vivo efficacy of quinazoline L-rhamnose derivatives.Acknowledgements
This research was supported by the Natural Science Foundation of China (Grant No. 81373322 and 21171154), the Key International Cooperation Project of CMST (Grant No. 2013DFG32160) and NSFC-Shandong Joint Fund (U1406402).Notes and references
- M. A. Olayioye, R. M. Neve, H. A. Lane and N. E. Hynes, EMBO J., 2000, 19, 3159–3167 CrossRef CAS PubMed.
- M. A. Lemmon and J. Schlessinger, Cell, 2000, 103, 211–225 CrossRef.
- N. E. Hynes and H. A. Lane, Nat. Rev. Cancer, 2005, 5, 341–354 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, N. Engl. J. Med., 2008, 358, 1160–1174 CrossRef CAS PubMed.
- D. S. Salomon, R. Brandt, F. Ciardiello and N. Normanno, Crit. Rev. Oncol. Hematol., 1995, 19, 183–232 CrossRef CAS PubMed.
- A. J. Barker, K. H. Gibson, W. Grundy, A. A. Godfrey, J. J. Barlow, M. P. Healy, J. R. Woodburn, S. E. Ashton, B. J. Curry, L. Scarlett, L. Henthorn and L. Richards, Bioorg. Med. Chem. Lett., 2001, 11, 1911–1914 CrossRef CAS PubMed.
- E. R. Wood, A. T. Truesdale, O. B. McDonald, D. Yuan, A. Hassell, S. H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, K. J. Alligood, D. W. Rusnak, T. M. Gilmer and L. Shewchuk, Cancer Res., 2004, 64, 6652–6659 CrossRef CAS PubMed.
- W. Pao and J. Chmielecki, Nat. Rev. Cancer, 2010, 10, 760–774 CrossRef CAS PubMed.
- G. R. Oxnard, M. E. Arcila, J. Chmielecki, M. Ladanyi, V. A. Miller and W. Pao, Clin. Cancer Res., 2011, 17, 5530–5537 CrossRef CAS PubMed.
- D. Li, L. Ambrogio, T. Shimamura, S. Kubo, M. Takahashi, L. R. Chirieac, R. F. Padera, G. I. Shapiro, A. Baum, F. Himmelsbach, W. J. Rettig, M. Meyerson, F. Solca, H. Greulich and K. K. Wong, Oncogene, 2008, 27, 4702–4711 CrossRef CAS PubMed.
- S. K. Rabindran, C. M. Discafani, E. C. Rosfjord, M. Baxter, M. B. Floyd, J. Golas, W. A. Hallett, B. D. Johnson, R. Nilakantan, E. Overbeek, M. F. Reich, R. Shen, X. Shi, H. R. Tsou, Y. F. Wang and A. Wissner, Cancer Res., 2004, 64, 3958–3965 CrossRef CAS PubMed.
- M. R. V. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. E. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. McFarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, J. Med. Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- L. V. Sequist, B. Besse, T. J. Lynch, V. A. Miller, K. K. Wong, B. Gitlitz, K. Eaton, C. Zacharchuk, A. Freyman, C. Powell, R. Ananthakrishnan, S. Quinn and J. C. Soria, J. Clin. Oncol., 2010, 28, 3076–3083 CrossRef CAS PubMed.
- J. B. Johnston, S. Navaratnam, M. W. Pitz, J. M. Maniate, E. Wiechec, H. Baust, J. Gingerich, G. P. Skliris, L. C. Murphy and M. Los, Curr. Med. Chem., 2006, 13, 3483–3492 CrossRef CAS PubMed.
- M. H. Potashman and M. E. Duggan, J. Med. Chem., 2009, 52, 1231–1246 CrossRef CAS PubMed.
- E. Leproult, S. Barluenga, D. Moras, J. M. Wurtz and N. Winssinger, J. Med. Chem., 2011, 54, 1347–1355 CrossRef CAS PubMed.
- C. H. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K. K. Wong, M. Meyerson and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 CrossRef CAS PubMed.
- W. Zhou, D. Ercan, L. Chen, C. H. Yun, D. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob, R. Padera, J. R. Engen, K. K. Wong, M. J. Eck, N. S. Gray and P. A. Janne, Nature, 2009, 462, 1070–1074 CrossRef CAS PubMed.
- P. Knesl, D. Roseling and U. Jordis, Molecules, 2006, 11, 286–297 CrossRef CAS PubMed.
- Z. Zhang, S. Wang, S. Wan, S. Ren, W. Li and T. Jiang, Carbohydr. Res., 2009, 344, 291–297 CrossRef CAS PubMed.
- V. Chandregowda, A. K. Kush and R. G. Chandrasekara, Eur. J. Med. Chem., 2009, 44, 3046–3055 CrossRef CAS PubMed.
- W. Li, Z. Zhang, S. Wang, S. Ren and T. Jiang, Chem. Biol. Drug Des., 2009, 74, 80–86 CAS.
- P. Wang, C. Li, J. Zang, N. Song, X. Zhang and Y. Li, Carbohydr. Res., 2005, 340, 2086–2096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06818a |
| This journal is © The Royal Society of Chemistry 2016 |