
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective ester hydrolysis by an achiral catalyst co-embedded with chiral amphiphiles into a vesicle membrane†
M.
Poznik
and
B.
König
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstraße 31, D-93053 Regensburg, Germany. E-mail: burkhard.koenig@chemie.uni-regensburg.de; Fax: +49 941 943 1717; Tel: +49 941 943 4575
First published on 29th April 2016
Abstract
Co-embedding of an amphiphilic non-chiral hydrolysis catalyst with amphiphilic chiral additives into the membrane of a phospholipid vesicle induces different rates of ester hydrolysis for enantiomeric amino acid esters.
The origin of chirality in nature,1 why natural molecules are homochiral, and how such a rapid amplification of single enantiomers occurred, are still unanswered questions.2,3 Synthetic chemistry uses chiral catalyst or ligands to convert achiral substrates enantio-selective.4 The reactant and the source of chirality require typical close contact. The simple addition of chiral additives to the reaction mixture or using enantiopure chiral solvents5,6 provide none or minute enantio-selectivity. The intermolecular chirality transfer improves in 3D networks, such as chiral MOFs7,8 or micellar solutions.9–12
The Raymond group reported an approach where chiral self-assembled capsules induce stereoselective reactions of achiral substrates by weak interactions.13–15
We report here the hydrolysis of enantiomeric amino acid esters on the modified surface of phospholipid vesicles (Fig. 1). A chiral, catalytically inactive membrane additive is co-embedded with a catalytically active achiral metal complex Zn2Cy into the phospholipid membrane.12,16 The membrane serves as two-dimensional platform with higher concentration of the amphiphilic membrane additives compared to the bulk solution.17 This proximity of the chiral additive to the achiral metal complex affects its selectivity in ester hydrolysis and induces thereby different reaction rates for both enantiomers.
 | ||
| Fig. 1 Proposed concept of additive-induced enantioselectivity in a hydrolytic reaction. | ||
The bis-zinc-cyclen complex Zn2Cy is anchored into the surface of the vesicle by its lipophilic alkyl chain and promotes the hydrolysis of activated carboxylic esters as previously reported (Fig. 2).18 The Lewis acidic zinc ions coordinate one water molecule and the ester functionality. Enantiopure amphiphilic derivatives of L-proline l-Pro, (−)-sparteine (−)-Spa, L-glucose l-Glu, L-tartaric acid l-Tar and L-histidine l-His-COOH and the corresponding alcohol l-His-OH were used as chiral membrane additives. Enantiometrically pure 4-nitrophenol esters of phenylalanine, PN-Phe, serve as substrates for the catalysed hydrolysis. Upon cleavage of the ester bond, the coloured 4-nitrophenolate anion is released, which enables a facile determination of the reaction progress. However, derivatives PN-Phe with free amine group show fast spontaneous hydrolysis under the reaction conditions. Therefore compounds PN-C12-Phe and PN-C2-Phe with a protected amino-group were prepared, which are stable in the absence of the hydrolysis catalyst.
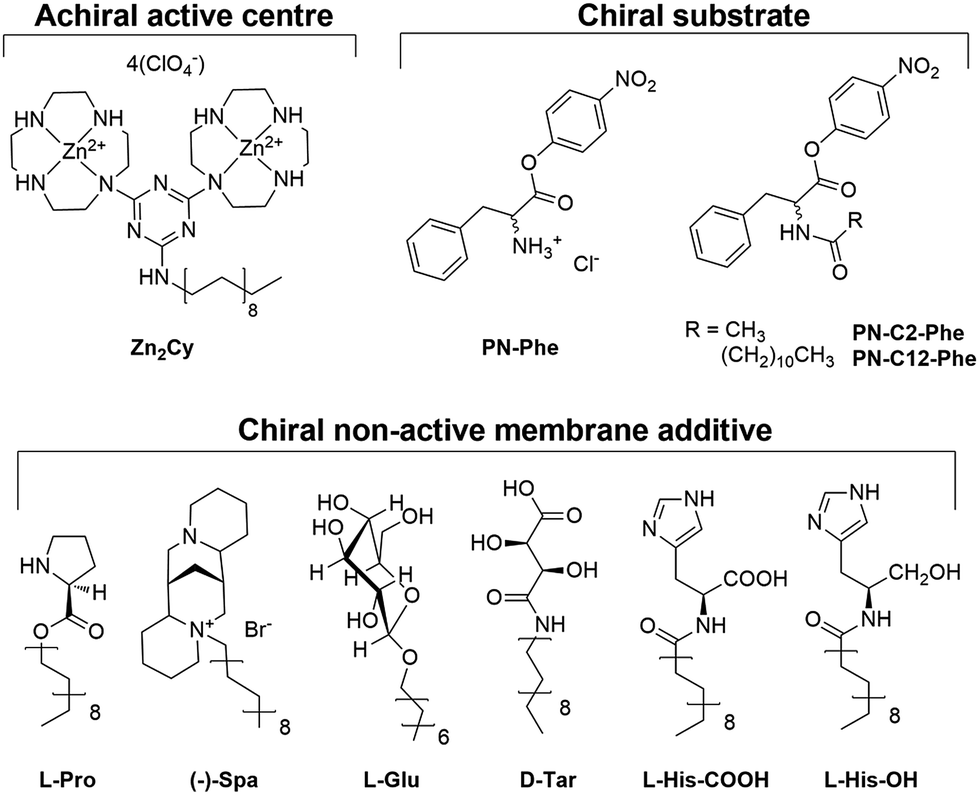 | ||
| Fig. 2 Molecular structures of the achiral catalyst for hydrolysis, racemic substrates and chiral membrane additives. | ||
Results and discussion
All measurements were done in buffered solutions (HEPES buffer, 25 mM, 7.4 pH), at room temperature. Samples were prepared by sonication according to a previously reported procedure to form micellar solutions or 100 nm unilamellar functionalised vesicles.18 The rate of substrate hydrolysis was determined colorimetrically as an increase in absorbance intensity at 400 nm (absorption maximum of 4-nitrophenol at pH 7.4; no other species absorb at this wavelength). The relative error of the measurement was estimated to be below 10% (Fig. 3). | ||
| Fig. 3 Examples of the kinetic data for hydrolysis of the PN-C12-Phe substrate by vesicular solution (85% DOPC), Zn2Cy (5%) and amphiphilic additive (10%). | ||
Pseudo first order rate constants were calculated using the initial slope method. Every membrane additive was examined as a sole micellar solution (without lipid or Zn2Cy), co-micellar solution (without lipid in the presence of Zn2Cy), and in vesicular membranes with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) lipids (5 mol% Zn2Cy, 10 mol% membrane additive, 85 mol% lipid). Lipids differ in their transition temperatures providing fluid (DOPC) and rigid (DSPC) membranes at room temperature. We measured the hydrolytic rate constants separately for the two enantiomers of the substrate PN-C12-Phe. This substrate is equipped with a long alkyl chain, which increases its lipophilicity and the adsorption to the surface of the membrane. The initial hydrolysis rates for different functionalized vesicles and co-micelles are summarised in Table 1.
| DOPC | DSPC | Co-micelles | No cyclen, no lipid | |||||
|---|---|---|---|---|---|---|---|---|
| k L | k D | k L | k D | k L | k D | k L | k D | |
| a No effect on hydrolysis observed. | ||||||||
| ZnCy2 | 31 | 30 | 65 | 50 | 201 | 198 | ||
| Pro | 15 | 15 | 63 | 61 | 166 | 159 | —a | —a |
| (−)-Spa | 28 | 26 | 50 | 41 | 277 | 236 | —a | —a |
| Glu | 13 | 15 | 28 | 28 | 229 | 242 | —a | —a |
| Tar | 24 | 14 | 26 | 24 | 46 | 46 | —a | —a |
| His-COOH | 212 | 113 | 153 | 129 | 214 | 286 | 11 | 2 |
| His-COOH | 127 | 233 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| His-OH | 339 | 327 | 357 | 287 | 513 | 384 | 6 | 4 |
Kinetic effects
Hydrolysis of the ester PN-C12-Phe is favoured in micellar solutions (Table 1). Confirming previous results,18 membrane additives can affect the hydrolysis activity in vesicular and co-micellar solutions significant.In DOPC membranes the hydrolytic rates are affected by two types of membrane additives: tartrate l-Tar decreases and histidine derivatives l-His-COOH and l-His-OH increase the initial rate of ester hydrolysis (Table 1). These observations are in accordance to effects on the previously studied hydrolysis of fluorescein diacetate by Zn2Cy.18
Enantiodiscrimination in ester hydrolysis
The rates of ester hydrolysis were determined for both enantiomers as previously reported for micellar solutions.10 The largest relative difference in hydrolysis rates for the enantiomeric esters is observed in DOPC vesicles with addition of amphiphilic tartrate l-Tar or histidine acid l-His-COOH as membrane additives (Fig. 4). | ||
| Fig. 4 Effect of membrane additives on the relative ratio of ester hydrolysis rates of enantiomeric amino acid esters represented by the fraction of pseudo first order kinetic constants. | ||
Both compounds contain free carboxy group. When the carboxyl group of histidine is reduced the effect in vesicles is lost. This indicates that the free acid might have a crucial role in this cooperative action. In buffered solution the acid is deprotonated and might interact with the Lewis acidic zinc complex. This interaction is weak in bulk solution, but may significantly increase due to the close proximity of the binding partners at the vesicular surface. In fluid DOPC membranes, embedded components diffuse and may thereby arrange optimal for the catalysis. In gel phase DSPC added amphiphiles form patches with restricted lateral movement decreasing the possibility for cooperative effects.19–21 This was also previously observed by us.18 Other membrane additives than tartrate or histidine do not show considerable effects on the relative hydrolysis rate of the enantiomeric esters. Substrates PN-C2-Phe and Pn-Phe exhibit similar enantio-selective enhancement (Table 2).
| k L [10−3 s−1] | k D [10−3 s−1] | |
|---|---|---|
| PN-C2-Phe | 35 | 17 |
| PN-Phe | 2059 | 1204 |
In order to confirm our observation, we prepared the enantiomeric amphiphilic histidine d-His-COOH. This compound was investigated under identical conditions as l-His-COOH and induced a similar hydrolysis rate enhancement of d-PN-C12-Phe (Fig. 5), but with opposite enantioselectivity.
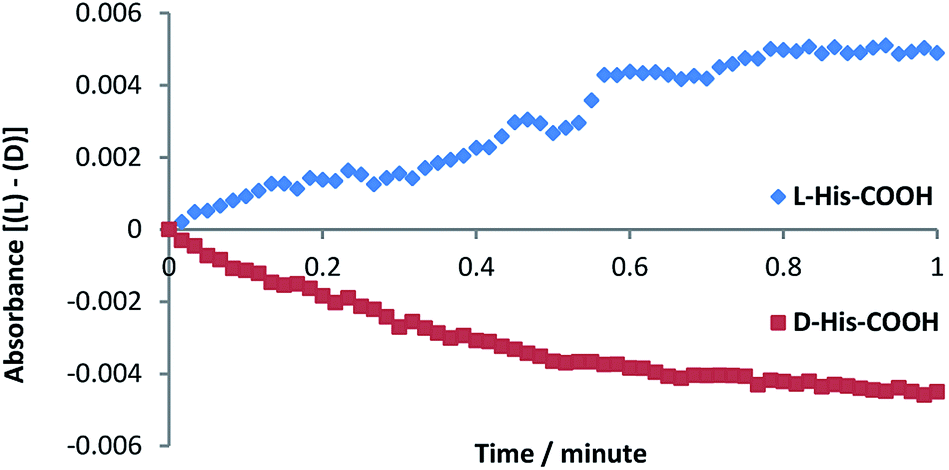 | ||
| Fig. 5 Recorded difference in kinetics of L and D PN-C12-Phe substrate hydrolysis by vesicular solution (85% DOPC) with L or D His-COOH (10%) and ZnCy2 (5%). | ||
In addition, the effect of the membrane additive loading on the enantioselectivity of the hydrolysis was investigated for d-His-COOH (Fig. 6). Addition of 10 mol% provided the highest relative difference between the hydrolysis rate constants.
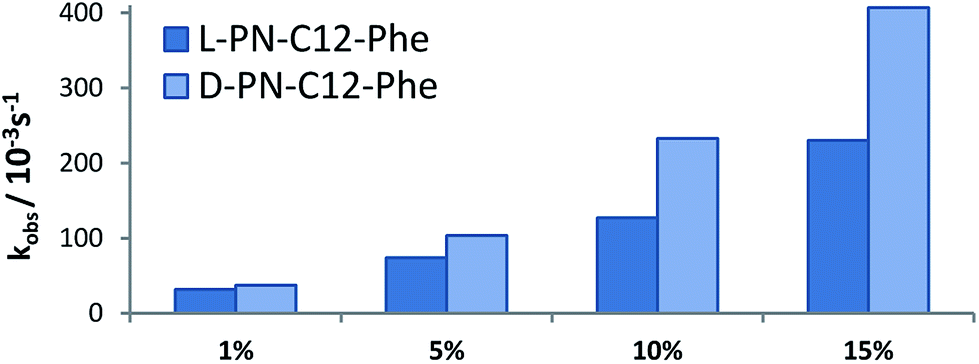 | ||
| Fig. 6 Pseudo first order rate constants of hydrolysis for both enantiomers by a vesicular solution of Zn2Cy (5 mol%) and DOPC with different loadings of d-His-COOH. | ||
Conclusions
The relative catalytic hydrolysis rates of enantiomeric amino acid esters with a non-chiral catalyst become significantly different, if the catalyst is co-embedded into the surface of DOPC vesicles with chiral amphiphiles. The rates for enantiomeric phenylalanine nitrophenyl esters differ by a factor of two with co-embedded amphiphiles prepared from tartaric acid or histidine. This resembles an ee of approx. up to 40%, which may be expected for reactions performed from racemate. The fluidity of the membrane is essential to achieve the catalytic enantio-discrimination and therefore only observed in DOPC vesicles. Although the measured rate differences may not be useful for practical applications, the results prove that the intermolecular interaction between a Lewis acidic metal complex, chiral amphiphiles and activated amino acid esters co-embedded into a fluid membrane without covalent connection can affect reaction rates enantioselective.Notes and references
- S. Mason, Trends Pharmacol. Sci., 1986, 7, 20–23 CrossRef CAS.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2010, 2, a002147 Search PubMed.
- W. Bonner, Origins Life Evol. Biospheres, 1991, 21, 59–111 CrossRef CAS.
- P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, Macmillan Education, 2008 Search PubMed.
- S. K. Tulashie, H. Lorenz, L. Hilfert, F. T. Edelmann and A. Seidel-Morgenstern, Cryst. Growth Des., 2008, 8, 3408–3414 CAS.
- S. K. Tulashie, H. Lorenz and A. Seidel-Morgenstern, Cryst. Growth Des., 2009, 9, 2387–2392 CAS.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680–2687 CrossRef PubMed.
- J. You, X. Yu, X. Li, Q. Yan and R. Xie, Tetrahedron: Asymmetry, 1998, 9, 1197–1203 CrossRef CAS.
- J.-S. You, X.-Q. Yu, X.-Y. Su, T. Wang, Q.-X. Xiang, M. Yang and R.-G. Xie, J. Mol. Catal. A: Chem., 2003, 202, 17–22 CrossRef CAS.
- R. A. Moss and W. L. Sunshine, J. Org. Chem., 1974, 39, 1083–1089 CrossRef CAS.
- F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Coord. Chem. Rev., 2009, 253, 2150–2165 CrossRef CAS.
- C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530–17531 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- C. Zhao, F. D. Toste, K. N. Raymond and R. G. Bergman, J. Am. Chem. Soc., 2014, 136, 14409–14412 CrossRef CAS PubMed.
- F. Mancin, P. Scrimin and P. Tecilla, Chem. Commun., 2012, 48, 5545–5559 RSC.
- B. Gruber and B. König, Chem.–Eur. J., 2013, 19, 438–448 CrossRef CAS PubMed.
- M. Poznik and B. Konig, Org. Biomol. Chem., 2014, 12, 3175–3180 CAS.
- E. J. Shimshick and H. M. McConnell, Biochemistry, 1973, 12, 2351–2360 CrossRef CAS PubMed.
- H.-J. Müller and H.-J. Galla, Eur. Biophys. J., 1987, 14, 485–491 CrossRef.
- F. A. Heberle and G. W. Feigenson, Cold Spring Harbor Perspect. Biol., 2011, 3, a004630 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06628c |
| This journal is © The Royal Society of Chemistry 2016 |