
l-Leucine supported on superparamagnetic silica-encapsulated γ-Fe2O3 nanoparticles: design, characterization, and application as a green catalyst for highly efficient synthesis of thiazoloquinolines
Abstract
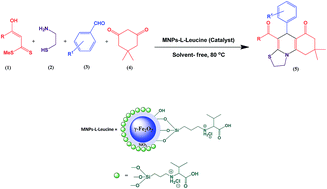
A new superparamagnetic silica-encapsulated γ-Fe2O3 supported L-leucine was successfully prepared and characterized by powder X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive spectrum (EDS), vibrating sample magnetometery (VSM), thermo-gravimetric analysis (TGA), and Fourier transform infrared spectroscopy (FT-IR). It catalyzed the synthesis of thiazoloquinolines through a four-component reaction of α-enolicdithioesters, cysteamine, aromatic aldehydes, and dimedone under thermal solvent-free conditions in good to excellent yields. This novel superparamagnetic catalyst could be easily separated from the reaction mixture with an external magnet and recovered at least five times without intense loss of catalytic activity.
 Please wait while we load your content...
Please wait while we load your content...

