
Polyaminocyclodextrin nanosponges: synthesis, characterization and pH-responsive sequestration abilities
Abstract
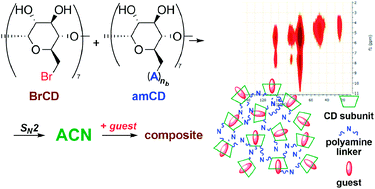
New pH-responsive nanosponges were obtained by reacting four different polyaminocyclodextrins with heptakis-(6-bromo)-(6-deoxy)-β-cyclodextrin. The materials obtained were characterized by various techniques (FT-IR, potentiometric titration, differential scanning calorimetry (DSC), porosimetry (BET), 13C{1H} CP-MAS NMR). Their adsorption abilities at different pH values were verified towards a suitable set of model guests, and seem mainly controlled by electrostatic interactions, as a function of the protonation/charge status of the polymer matrix. By contrast, data positively point out a lesser importance assumed by the induced-fit effect, important in affecting the formation of host–guest complexes in solution. The frequency-switched Lee-Goldburg (FSLG) heteronuclear correlation solid-state NMR technique was exploited in order to assess the possible location of the guests within the polymer matrix.
 Please wait while we load your content...
Please wait while we load your content...

