
A ruthenium derivative of quercetin with enhanced cholesterol-lowering activity

Abstract
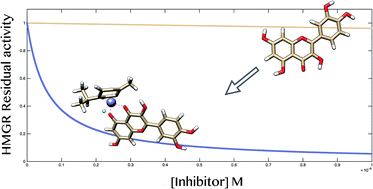
A ruthenium(II) p-cymene derivative of quercetin was synthesized and functionally tested for cholesterol-lowering ability via direct 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR) inhibition. Ruthenium complexation dramatically increased the inhibition potency of the parent quercetin toward HMGR, with a consequent enhancement of the cholesterol-lowering effect in hepatic cells.
 Please wait while we load your content...
Please wait while we load your content...

