
ADMET polymerization of biobased monomers deriving from syringaresinol†
Louis Hollande‡
a,
Abdus Samad Jaufurally‡ab,
Paul-Henri Ducrot*b and
Florent Allais*ac
aAgroParisTech, Chaire Agro-Biotechnologies Industrielles (ABI), CEBB – 3 rue des Rouges Terres, Route de Bazancourt, F-51110 Pomacle, France. E-mail: florent.allais@agroparistech.fr
bInstitut Jean-Pierre Bourgin, INRA, AgroParisTech, CNRS, Université Paris-Saclay, RD10, 78026 Versailles Cedex, France. E-mail: ducrot@versailles.inra.fr
cUMR 782 GMPA, INRA, AgroParisTech, CNRS, Université Paris-Saclay, Avenue Lucien Brétignières, F-78850 Thiverval-Grignon, France
First published on 27th April 2016
Abstract
Renewable α,ω-dienes have been prepared from syringaresinol, a naturally occurring bisphenol deriving from sinapyl alcohol, and further studied as monomers in ADMET polymerizations. Polymerization was optimized according to catalyst loading and reaction conditions (in mass vs. in solvent), and led to polymers with molecular weight up to 14.1 kDa. Thermal analyses of these new polymers showed excellent thermal stabilities (257–360 °C) and tunable Tg (18–70 °C) depending on the structure of the starting α,ω-diene monomer.
Introduction
The collapse of fossil resources and rising prices may have been the first trigger for a biobased economy. Nevertheless, today, industries, customers and regulators (e.g., REACH) are increasingly demanding for eco-friendly and bio-based chemicals issued from sustainable industrial processes. In this context, the production of new platform chemicals from biomass through green processes is an alluring strategy for a sustainable development. Indeed, biomass offers a wide range of molecules that can be used to access valuable synthons such as polyols, furans, fatty acids, aliphatic alkanes/alkenes or aromatics/phenolics to name a few.1 Out of these sustainable and valuable chemical feedstocks, sinapic acid, from the ancient greek σιναπι (sinapi), a major compound isolated from Brassicaceae seeds and also one of the three major p-hydroxycinnamic acids found in lignocellulosic biomass, is one of the most promising substitute to fossil phenolics. It is present in relatively large quantities in the form of sinapine (choline ester of sinapic acid) in the seeds of oil plants from the Brassicaceae family.2In order to offer some new opportunities of valorization of this compound, and having dedicated ourselves to the synthesis and valorization of lignin-derived phenolics and more particularly p-hydroxycinnamic acids and derivatives,3–14 we recently optimized a chemo-enzymatic synthetic pathway allowing the synthesis of syringaresinol from sinapic acid (or syringaldehyde) in very high yield and purity.15 The work reported here presents the use of syringaresinol as precursor for the synthesis of novel α,ω-dienes monomers incorporating the two aromatic rings of syringaresinol linked through a cis-fused bisfuranic moiety. These monomers were then submitted to acyclic diene metathesis (ADMET) polymerization.
Indeed, thanks to the ease of handling and high functional group tolerance of the Ru-based catalysts used in ADMET, this method is a very useful approach for the construction of well defined polymer architectures16 and allowed the synthesis of renewable polymers, such as polyesters, polyethers, polyamides and many others, very promising for commercial applications.12,17–27 In spite of extensive research to develop commercial ferulic acid-,12,17 eugenol-,22 vanillin-23 and bisvanillin-24 renewable polymers, to the best of our knowledge, there is no example of ADMET polymerization involving sinapic acid derivatives (i.e., syringaresinol, syringaldehyde) as diene substrates.
The structure and thermal properties of these novel polymers were thereafter studied in order to evaluate their potential in industrial applications.
Experimental
Materials and methods
All reagents were purchased from Sigma-Aldrich or Tokyo Chemical Industry Co and used as received. Solvents were purchased from ThermoFisher Scientific, DMF was dried on a mBraun SPS 800 system. Deuterated chloroform (CDCl3) was purchased from Euriso-top. Evaporations were conducted under reduced pressure at temperature below 40 °C. Column chromatographies were carried out with an automated flash chromatography (PuriFlash 4100, Interchim) and pre-packed INTERCHIM PF-30SI-HP (30 μm silica gel) columns. FT-IR and UV analyses were performed on Cary 630 FTIR and Cary 60 UV-Vis from Agilent technologies, respectively. NMR analyses were recorded on a Bruker Fourier 300. 1H NMR spectra of samples were recorded in CDCl3 at 300 MHz (residual CHCl3 signal at δ = 7.26 ppm). 13C NMR spectra of samples were recorded at 75 MHz (CDCl3 signal at δ = 77.16 ppm). Thermo-gravimetric analyses (TGA) were recorded on a Q500, from TA. Around 5 mg of each sample was heated at 10 °C min−1 from 50 to 500 °C under nitrogen flow (60 mL min−1). Differential scanning calorimetry (DSC) thermograms were obtained using a DSC Q20, from TA, under inert atmosphere (N2). Around 5 mg were weighed in a pan which was then sealed and submitted to the following heat/cool/heat cycle: equilibration at −60 °C and hold for 5 min, heating from −60 °C to 200 °C at 10 °C min−1 and hold for 5 min, cooling from 200 °C to −60 °C at 10 °C min−1. Gel Permeation Chromatography (GPC) was performed at 40 °C on an Infinity 1260 system from Agilent Technologies with a quadruple detection (IR, UV, MALS, viscosimetry) and two PL-gel 5 mm mixed D column (300 mm × 7.5 mm) in THF (flow rate 1 mL min−1) using polystyrene calibration. HRMS were recorded by the PLANET platform at URCA on a Micromass GC-TOF.Synthesis of (+/−)-syringaresinol
Sinapyl alcohol (4 g, 19 mmol, 1 eq.) was dissolved in acetonitrile (80 mL) in a triple-neck round bottom flask equipped with a cooling system and citrate/phosphate buffer (320 mL) at pH 5 were added (0.05 M). A solution containing 35.2 mg of laccase from Trametes versicolor (0.1 U mg−1 of substrate) in 50 mL of citrate/phosphate buffer was added dropwise to the monolignol solution at a rate of 9 mL per hour with a syringe pump. The reaction mixture was stirred with a magnetic stirrer in the presence of O2 (air) at 50 °C for 470 min in darkness. At the end of the reaction, the product was extracted with dichloromethane (2 × 150 mL) and ethyl acetate (2 × 150 mL). The organic phases were combined, dried over anhydrous MgSO4, filtered and the solvent was evaporated under reduced pressure and no further purification was needed.White powder (93%). Mp: 169 °C. FT-IR (neat): νmax 3424 (OH), 1400–1600 (Ar), 1000–1300 (C–O–C). HRMS: (TOF MS, ES+): m/z calcd for C22H26O8Na: 441.1525; found: 441.1516. 1H NMR (300 MHz, CDCl3, δ): 3.10 (m, 2H, Hβ), 3.90 (s, 12H, H5), 3.90 (m, 2H, Hγ), 4.27 (dd, 2H, J = 6.9 Hz and 9.0 Hz, Hγ′), 4.73 (d, 2H, J = 4.2 Hz, Hα), 5.51 (s, 2H, Hphenol), 6.59 (s, 4H, H2). 13C NMR (75 MHz, CDCl3, δ): 54.4 (Cβ), 56.4 (C5), 71.8 (Cγ), 86.1 (Cα), 102.7 (C2), 132.1 (C1), 134.3 (C4), 147.2 (C3).
General procedure for the synthesis of α,ω-diene monomers
Syringaresinol (1 equiv.) and K2CO3 (5 equiv.) were dissolved in dry DMF (2 M) under nitrogen. Bromo-alkene (4 equiv.) was then added, and the mixture was stirred and heated at 80 °C for 12 h. Reaction was quenched with water (v(H2O)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) v(DMF)) and the aqueous layer was extracted three times with ethyl acetate (3 × v(H2O)). Organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Crude product was purified by flash chromatography on silica gel cyclohexane
v(DMF)) and the aqueous layer was extracted three times with ethyl acetate (3 × v(H2O)). Organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Crude product was purified by flash chromatography on silica gel cyclohexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOEt (from 95/5 to 50/50) to yield the corresponding α,ω-diene monomer.
:AcOEt (from 95/5 to 50/50) to yield the corresponding α,ω-diene monomer.
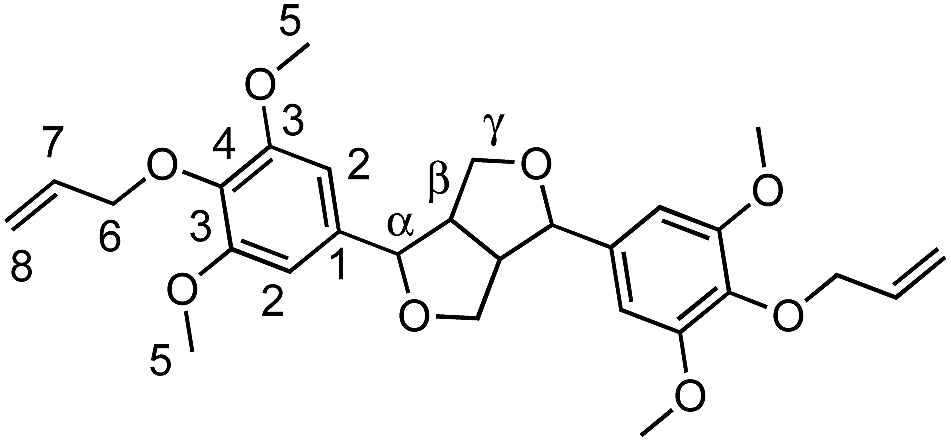
White powder (75%). FTIR (neat): νmax: 1501 (CC Ar). HRMS: (TOF MS, ES+): m/z calcd for C28H34O8Na [MNa]+: 521.2151; found: 521.2142. 1H NMR (300 MHz, CDCl3, δ): 3.10 (m, 2H, Hβ), 3.85 (s, 12H, H5), 3.91 (dd, 2H, J = 3.6 Hz, Hγ), 4.33 (m, 2H, Hγ′), 4.48 (d, 4H, J = 7 Hz, H6), 4.73 (d, 2H, J = 4 Hz, Hα), 5.15 (m, 2H, H8), 5.32 (m, 2H, H8), 6.10 (m, 2H, H7), 6.58 (s, 4H, H2). 13C NMR (75 MHz, CDCl3, δ): 54.6 (Cβ), 57.3 (C5), 71.9 (C6), 74.2 (Cγ,γ′), 85.9 (Cα), 102.87 (C2), 117.7 (C8), 134.5 (C7), 136.1 (C1), 136.7 (C4), 153.63 (C3).
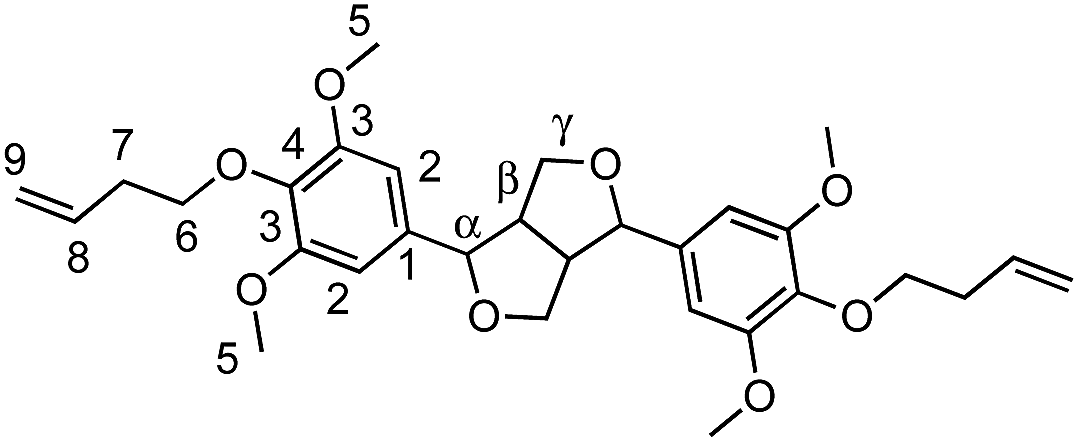
Viscous oil (78%). FTIR (neat): νmax: 1501 (CC, Ar). HRMS: (TOF MS, ES+): m/z calcd for C30H38O8Na [MNa]+: 549.2464; found: 549.2474. 1H NMR (300 MHz, CDCl3, δ): 2.49 (q, 4H, J = 6.9 Hz and 13.8 Hz, H7), 3.10 (m, 2H, Hβ), 3.84 (s, 12H, H5), 3.90 (m, 2H, J = 3.6 Hz, Hγ), 3.99 (t, 4H, J = 7.2 Hz, H6), 4.30 (m, 2H, Hγ′), 4.72 (d, 2H, J = 4 Hz, Hα), 5.09 (m, 4H, H9), 5.87 (m, 2H, H8), 6.6 (s, 4H, H2). 13C NMR (75 MHz, CDCl3, δ): 34.5 (C7), 54.3 (Cβ), 56.2 (C5), 71.9 (C6), 72.4 (Cγ,γ′), 86.0 (Cα), 102.9 (C2), 116.4 (C9), 134.9 (C8), 136.6 (C4,1), 153.6 (C3).
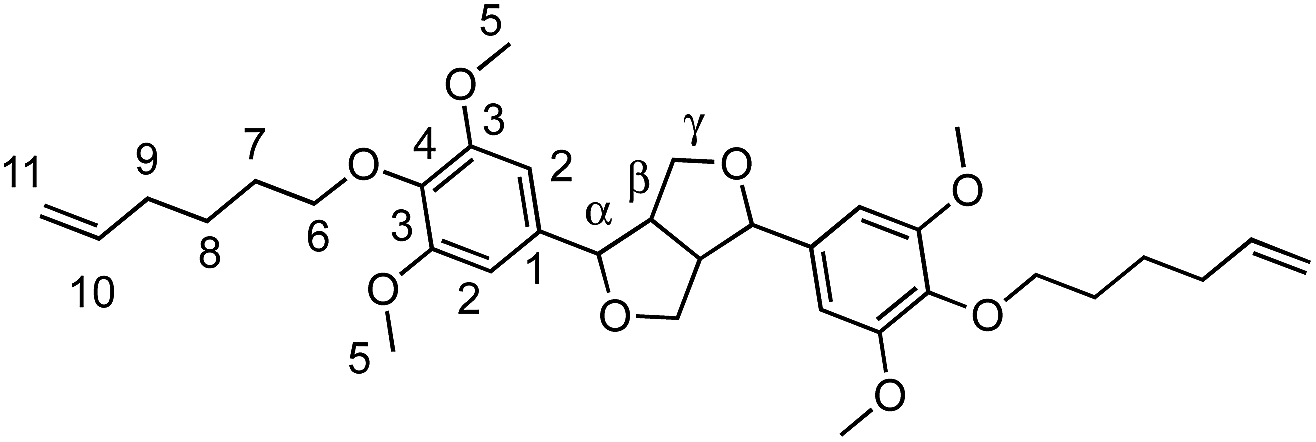
Viscous oil (71%). FTIR (neat): νmax: 1501 (CC Ar). HRMS: (TOF MS, ES+): m/z calcd for C34H46O8Na [MNa]+: 605.3090; found: 605.3098. 1H NMR (300 MHz, CDCl3, δ): 1.54 (m, 4H, H8), 1.7 (qt, 4H, J = 6.9 Hz, H7), 2.1 (q, 4H, J = 7.2 Hz, H9), 3.11 (m, 2H, Hβ), 3.84 (s, 12H, H5), 3.84 (m, 2H, Hγ), 3.93 (m, 4H, H6), 4.31 (m, 2H, Hγ′), 4.73 (d, 2H, J = 4.2 Hz, Hα), 4.94–5.04 (m, 4H, H11), 5.84 (m, 2H, H10), 6.58 (s, 4H, H2). 13C NMR (75 MHz, CDCl3, δ): 25.1 (C8), 29.5 (C7), 33.5 (C9), 54.3 (Cβ), 56.2 (C5), 71.9 (C6), 73.21 (Cγ,γ′), 86.0 (Cα), 103.0 (C2), 114.41 (C11), 136.4 (C1), 136.8 (C4), 138.9 (C10), 153.7 (C3).
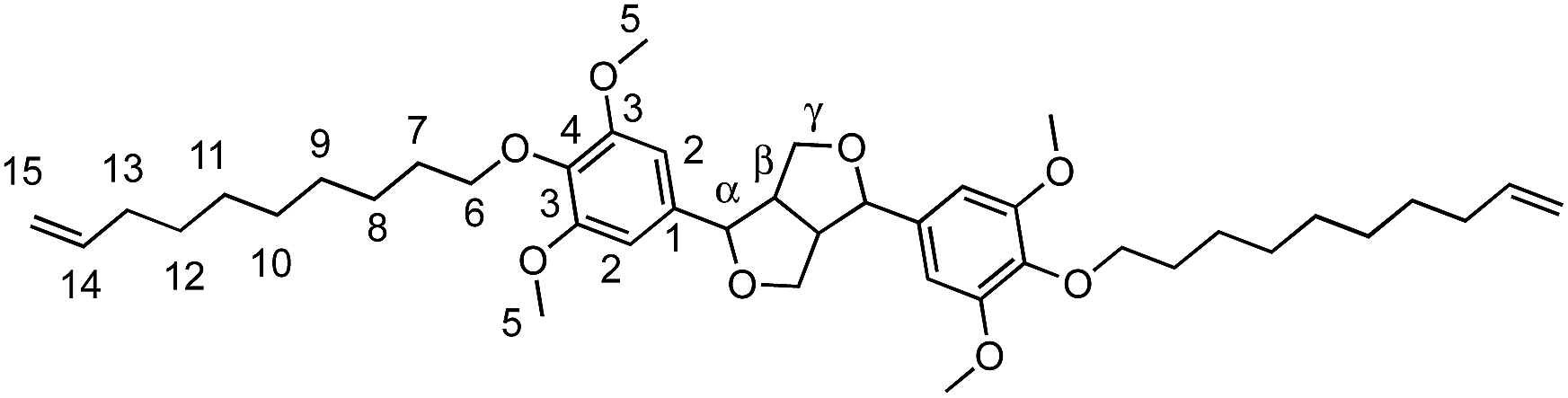
Viscous oil (77%). FTIR (neat): νmax: 1502 (CC Ar). HRMS: (TOF MS, ES+): m/z calcd for C42H62O8Na [MNa]+: 717.4342; found: 717.4351. 1H NMR (300 MHz, CDCl3, δ): 1.30–1.44 (m, 20H, H8–12), 1.73 (qt, 4H, J = 7.8 Hz, H7), 2.02 (q, 4H, J = 7.1 Hz, H13), 3.1 (m, 2H, Hβ), 3.83 (s, 12H, H5), 3.92 (m, 6H, Hγ,6), 4.29 (m, 2H, Hγ′), 4.72 (d, 2H, J = 4.2 Hz, Hα), 4.89–5.28 (m, 4H, H15), 5.75 (m, 2H, H14), 6.55 (s, 4H, H2). 13C NMR (75 MHz, CDCl3, δ): 25.8 (C8), 28.9–30.1 (C7,9–12), 33.8 (C13), 54.3 (Cβ), 56.1 (C5), 71.9 (C6), 73.51 (Cγ,γ′), 86.0 (Cα), 103.0 (C2), 114.1 (C15), 136.3 (C1), 136.9 (C4), 139.2 (C14), 153.7 (C3).
General procedure for ADMET polymerizations
Results and discussion
α,ω-Diene monomers SYR-All, SYR-But, SYR-Hex and SYR-Dec were prepared by conducting Williamson etherification of syringaresinol with four bromo-alkenes (3-bromoprop-1-ene, 4, bromobut-1-ene, 6-bromohex-1-ene and 10-bromodec-1-ene) in DMF at 80 °C, for 12 hours under nitrogen, in presence of potassium carbonate (Scheme 1). These four α,ω-diene monomers were used to study the impact of the chain length of the olefin on the polymerizations and the thermal properties of the resulting polymers.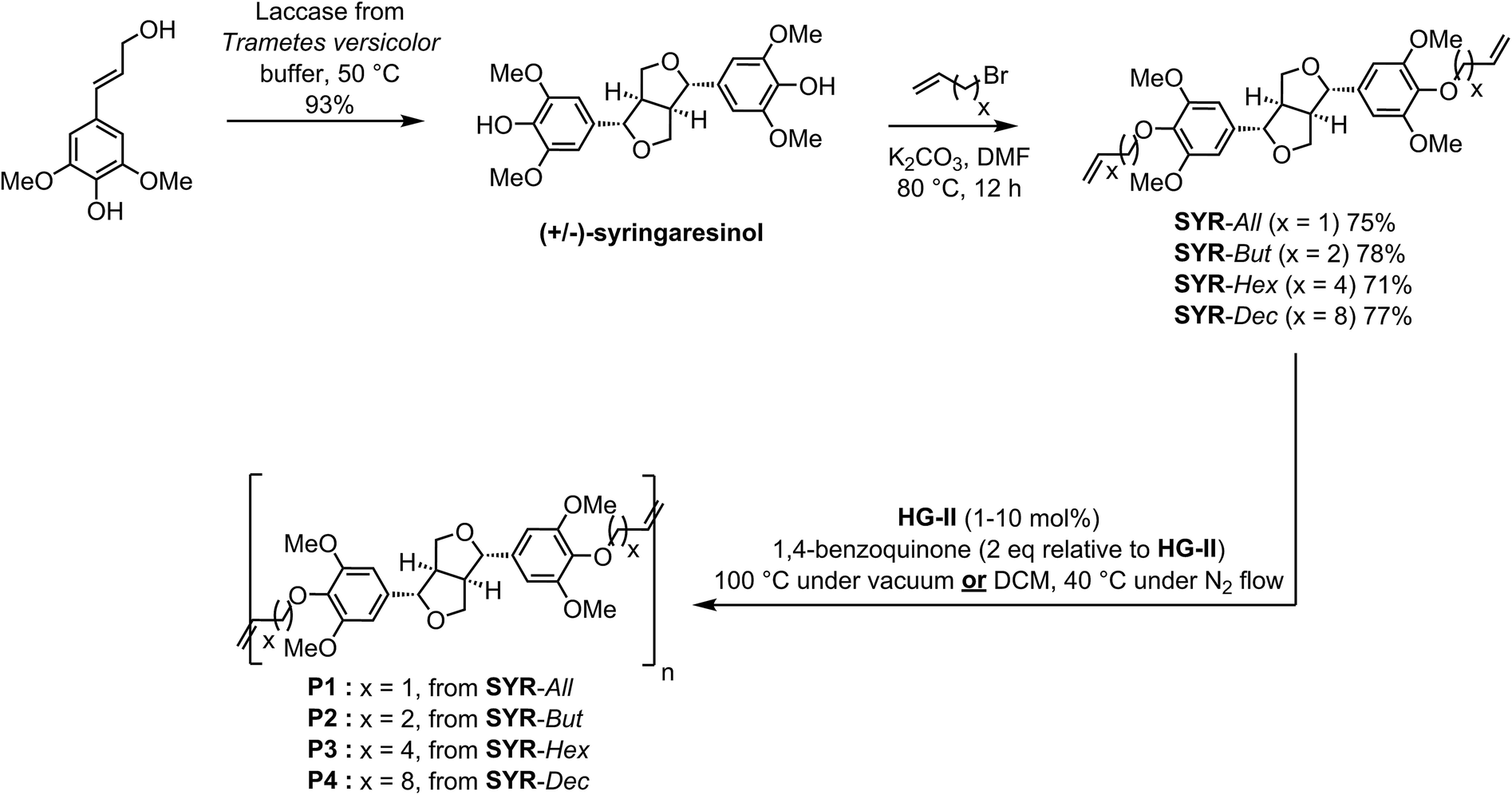 | ||
| Scheme 1 Synthesis and ADMET polymerization of the four syringaresinol derived α,ω-diene monomers. | ||
ADMET polymerization
We first studied the reactivity of the four syringaresinol-derived α,ω-diene in ADMET polymerization. Though it has been reported that ADMET polymerizations are favored when carried out under bulk conditions (aka in mass),12,28 we also performed polymerizations in solvent to verify if this also applied to syringaresinol-based α,ω-diene monomers (Scheme 1). For ADMET reactions conducted in mass at 100 °C, continuous vacuum (ca. 20 mbar) was applied to guarantee the efficient removal of ethylene and thus shift the equilibrium towards polymerization. The latter temperature was chosen as it has been already shown that, in the ADMET polymerization of rigid IDF-based α,ω-dienes12 incorporating two dihydroferulate esters of isosorbide, heating the reaction mixture at 100 °C significantly improves the molecular weights by lowering the viscosity of the reaction medium thus facilitating both ethylene removal and stirring. Dichloromethane, previously reported as a suitable solvent for ADMET,29 was used for in solvent polymerizations. Solvent reactions were performed at 40 °C under a low and steady nitrogen flow to remove ethylene but not dichloromethane. In both reaction conditions, 1,4-benzoquinone (2 eq. relative to catalyst) was used to limit olefin isomerization.30 Finally, Hoveyda–Grubbs second generation catalyst (HG-II), with loadings varying from 0.1 to 10 mol%, was the only catalyst tested in this study as previous works by Abbas,31 Firdaus23 and Barbara12 have shown HG-II to be the most active towards the cross metathesis of acrylates-, vanillin- and ferulic acid-based α,ω-dienes, respectively. All ADMET reactions were run for 4 hours and followed by GPC in order to identify the best conditions (nature of the α,ω-diene monomer, in mass vs. in solvent, catalyst loading) (Fig. 2). Table 1 summarizes the results of these polymerizations. The 1H and 13C NMR spectra of all polymers are displayed in the ESI.†| Entry | ADMET conditions | HG-II (mol%) | Monomer | Polymer | Mn (kDa) | Mw (kDa) | DPn | ĐM | Conversionc (%) | Td5%d (°C) | Td50%d (°C) | Tge (°C) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a HG-II (1, 5 or 10 mol%), 1,4-benzoquinone (2 equivalent relative to HG-II), DCM (2 M), 40 °C, 4 hours.b HG-II (1, 5 or 10 mol%), 1,4-benzoquinone (2 equivalent relative to HG-II), vacuum, 100 °C, 4 hours.c Calculated with the residual monomer amounts determined from the corresponding peak area on SEC traces of crude reaction mixtures prior precipitation.d TGA data recorded at 10 °C min−1 under nitrogen (60 mL min−1).e DSC data recorded at 10 °C min−1 under nitrogen (60 mL min−1), value determined at the 2nd heating scan. | ||||||||||||
| 1 | In massb | 1 | SYR-All | P1 | 2.0 | 2.6 | 4 | 1.3 | 59 | 283 | 352 | 65 |
| 2 | In massb | 5 | SYR-All | 2.2 | 2.9 | 4 | 1.3 | 73 | 266 | 327 | 70 | |
| 3 | In massb | 10 | SYR-All | 2.8 | 3.9 | 6 | 1.4 | 80 | 263 | 343 | 68 | |
| 4 | In solventa | 1 | SYR-All | P1 | 1.4 | 1.6 | 2 | 1.1 | — | — | — | — |
| 5 | In solventa | 5 | SYR-All | 1.6 | 2.1 | 3 | 1.3 | — | — | — | — | |
| 6 | In solventa | 10 | SYR-All | 2.2 | 2.8 | 4 | 1.2 | — | — | — | — | |
| 7 | In massb | 1 | SYR-But | P2 | 4.5 | 7.7 | 9 | 1.7 | 93 | 283 | 376 | 58 |
| 8 | In massb | 5 | SYR-But | 5.1 | 8.5 | 10 | 1.7 | 88 | 274 | 369 | 60 | |
| 9 | In massb | 10 | SYR-But | 6.7 | 13.1 | 13 | 1.9 | 89 | 272 | 339 | 61 | |
| 10 | In massb | 1 | SYR-Hex | P3 | 8.9 | 18.3 | 15 | 2.1 | 94 | 308 | 392 | 39 |
| 11 | In massb | 5 | SYR-Hex | 8.3 | 15.5 | 14 | 1.9 | 90 | 268 | 342 | 40 | |
| 12 | In massb | 10 | SYR-Hex | 8.9 | 16.8 | 15 | 1.9 | 86 | 257 | 324 | 38 | |
| 13 | In solventa | 1 | SYR-Hex | P3 | 1.4 | 1.5 | 2 | 1.1 | — | — | — | — |
| 14 | In solventa | 5 | SYR-Hex | 8.4 | 30.0 | 14 | 3.6 | — | — | — | — | |
| 15 | In solventa | 10 | SYR-Hex | 8.2 | 26.1 | 14 | 3.2 | — | — | — | — | |
| 16 | In massb | 1 | SYR-Dec | P4 | 13.0 | 25.5 | 19 | 2.0 | 97 | 360 | 393 | 20 |
| 17 | In massb | 5 | SYR-Dec | 12.7 | 28.6 | 19 | 2.2 | 95 | 349 | 392 | 19 | |
| 18 | In massb | 10 | SYR-Dec | 14.1 | 32.0 | 21 | 2.3 | 82 | 347 | 394 | 18 | |
Structural analysis
In addition to GPC, 1H NMR spectrometry of the resulting crude polymerization mixtures was also performed not only to confirm the polymers structures but also to reveal potential undesired side reactions such as ring-closing metathesis (RCM) or olefin isomerization (Fig. 1). 1H NMR spectra of monomers (SYR-All (A), SYR-But (B), SYR-Hex (C), SYR-Dec (D)) and their corresponding polymers obtained in mass reveal the decrease of intensity of the signals at 5.0–5.2 and 5.8–5.9 ppm corresponding to the terminal olefin protons along with the formation of the internal double bound whose signal is at 5.5 ppm (see ESI†). ADMET polymerizations performed in solvent led to short oligomers (Table 1, entries 4–6 and 13–15) and higher dispersity (Table 1, entries 14 and 15), whereas in mass ADMET polymerizations gave higher molecular weights as proven by the GPC results (Table 1, Fig. 2). Detailed analyses of 1H and 13C NMR spectra show no evidence of isomerization of the double bond (ESI†). | ||
| Fig. 1 1H NMR spectra of SYR-All, SYR-But, SYR-Hex and SYR-Dec and their corresponding oligomers and polymers (P1, P2, P3 and P4). | ||
 | ||
| Fig. 2 GPC traces of P1, P2, P3 and P4 (10 mol% catalyst). | ||
On the basis of the results reported in Table 1 and Fig. 1, it was concluded that in mass procedure with 1 mol% HG-II, at 100 °C for 4 hours was the best procedure for ADMET polymerization for all monomers. In such conditions, syringaresinol-based polymers were obtained with Mn in the range of 4.5–14.1 kDa.
It is also noteworthy that ADMET polymerization of monomers bearing allyl moieties only provides low molecular weight oligomers. Furthermore, no RCM product was observed in the case of SYR-All, probably because of the ring strain of syringaresinol that prevents intramolecular ring closing metathesis.
Because in solvent ADMET conditions proved unsuitable for our monomers, only results for polymers obtained in mass are being considered in the following discussion (Table 1, entries 1–3, 7–12 and 16–18). As previously observed in the case of isosorbide based α,ω-dienes,12 the rigid bicyclic structure of syringaresinol and its relatively high viscosity restricts the polymerization to a degree of polymerization (DPn) up to ca. 20 (Table 1, entries 16–18). As expected, because of decreasing viscosity, the longer the α,ω-dienes the higher the Mn (Table 1, entries 3, 9, 12 and 18). Interestingly, while molecular weights (Mn) of polymers increase by 98% when but-1-ene was replaced by hex-1-ene (P3 vs. P2, Table 1, entries 10 vs. 7), they increase only by 46% when hex-1-ene is replaced by dec-1-ene (P4 vs. P3, Table 1, entries 16 vs. 10). Increasing the catalyst loading above 1 mol% resulted in similar to slightly higher molecular weights. In the other hand, lower catalyst loadings (e.g., 0.1–0.5 mol%) did not lead to higher molecular weight, neither did longer reactions times (8 hours vs. 4 hours).
In terms of reactivity, syringaresinol-based α,ω-diene proves as efficient as recently published IDF-Hex and IDF-Dec α,ω-dienes which give similar molecular weights when submitted to ADMET polymerization with HG-II (Scheme 2).12
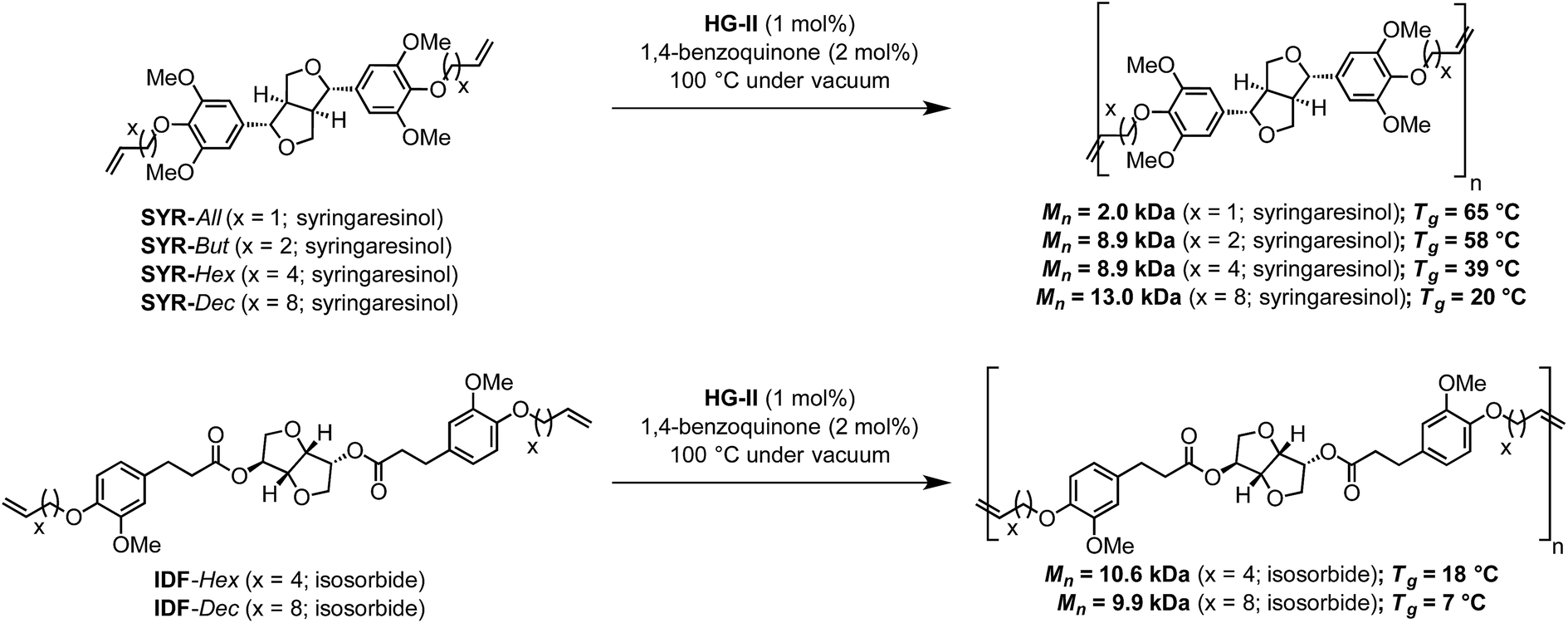 | ||
| Scheme 2 Syringaresinol- and ferulic acid-derived12 polymers via ADMET. | ||
Thermal properties
The thermal properties of monomers and polymers described in Table 1 were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC).TGA analyses of the monomers revealed a thermostability (Td50%) in the range of 333–392 °C (Table 2). Furthermore, the nature of the alkene impacts the degradation temperature (Td50%). Indeed replacing but-1-ene by dec-1-ene strongly increases their thermostability by 58 °C (Table 2).
| Monomer | SYR-All | SYR-But | SYR-Hex | SYR-Dec |
|---|---|---|---|---|
| Td5% (°C) | 220 | 273 | 300 | 328 |
| Td50% (°C) | 333 | 334 | 358 | 392 |
Thermal analyses of polymers P1–P4 showed significant differences in Td5% and glass transition temperature (Tg) depending on the alkene length (C4, C6 and C10) (Fig. 3 & 4). Because of the presence of aromatic moieties and the rigidity of syringaresinol, all polymers exhibit a thermostability in the range of 257–360 °C (Td5%), the dec-1-ene based ones being the most stable (Table 1, entries 16, 17 and 18).
 | ||
| Fig. 3 TGA analyses for polymers P1, P2, P3, and P4 (under nitrogen, 10 °C min−1). | ||
 | ||
| Fig. 4 DSC analyses of P1, P2, P3 and P4 (2nd heating cycle, under nitrogen, 10 °C min−1). | ||
DSC analyses also reveal that a decrease of the alkene chain length (from decene to butene) results in an inversely proportional increase of the Tg (P4 < P3 < P2 < P1). Varying the nature of the α,ω-diene monomers thus provides Tg in the range of 18 to 70 °C (Fig. 4 & 5). Compared to IDF-Hex α,ω-diene monomer-based polymer12 that exhibits a Tg of 18 °C, that of P3 deriving from SYR-Hex monomer is of 39 °C (Scheme 2). Such an increase could be explained by not only the shorter distance between the two phenols in the α,ω-diene monomer (12 atoms vs. 4 carbon atoms), but also by the presence of extra methoxy groups on the aromatic rings that increases the conformational barriers for chain motion.32 Finally, it is noteworthy to mention that all polymers are amorphous and do not show melting points (Tm) on DSC.
 | ||
| Fig. 5 Alkene chain-length dependence of the glass transition temperature (Tg). | ||
Conclusions
In summary, syringaresinol-based α,ω-diene monomers were obtained in very good yields through a chemo-enzymatic synthetic pathway and were successfully polymerized via ADMET in presence of second generation Hoveyda–Grubbs catalyst. Mass polymerization resulted in polymers with Mn as high as 14.1 kDa. Thermal analyses through TGA and DSC demonstrated that these polymers are thermostable up to 257–360 °C and, more importantly, that their Tg can be easily tuned by adjusting the alkene length of the α,ω-diene monomers (from 18 to 70 °C). Prepared from renewable feedstocks, these polymers could be envisaged as sustainable substitutes to conventional petro-based polyesters.Acknowledgements
The authors are grateful to the Region Champagne-Ardenne, the Conseil Général de la Marne and Reims Métropole for their financial support.References AND notes
- (a) J. J. Bozell, J. O. Hoberg, et al., in Green Chemistry – Frontiers in Benign Chemical Synthesis and Processes, Oxford University, 1998, pp. 27–45 Search PubMed; (b) C. Okkerse and H. van Bekkum, Green Chem., 1999, 1, 107 RSC; (c) D. L. Klass, in Biomass for Renewable Energy, Fuels, and Chemicals, Academic Press, 1998, pp. 495–542, 91–157 Search PubMed.
- N. Niciforovic and H. Abramovi, Compr. Rev. Food Sci. Food Saf., 2014, 13, 34–51 CrossRef CAS.
- M. Quentin, V. Allasia, A. Pegard, F. Allais, P.-H. Ducrot, B. Favery, C. Levis, S. Martinet, C. Masur, M. Ponchet, D. Roby, L. Schlaich, L. Jouanin and H. Keller, PLoS Pathog., 2009, 5(1) DOI:10.1371/journal.ppat.1000264.
- F. Allais, M. Aouhansou, A. Majira and P.-H. Ducrot, Synthesis, 2010, 16, 2787 CrossRef.
- F. Allais and P.-H. Ducrot, Synthesis, 2010, 16, 1649 CrossRef.
- L. M. M. Mouterde, A. L. Flourat, M. M. M. Cannet, P.-H. Ducrot and F. Allais, Eur. J. Org. Chem., 2013, 1, 173 CrossRef.
- B. Cottyn, A. Kollman, P. Waffo Teguo and P.-H. Ducrot, Chem.– Eur. J., 2011, 17, 7282 CrossRef CAS PubMed.
- (a) J. C. Dean, R. Kusaka, P. S. Walsh, F. Allais and T. S. Zwier, J. Am. Chem. Soc., 2014, 136, 14780 CrossRef CAS PubMed; (b) L. A. Baker, M. D. Horbury, S. E. Greenough, F. Allais, P. S. Walsh, S. Habershon and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 56–61 CrossRef CAS PubMed.
- F. Pion, A. F. Reano, P.-H. Ducrot and F. Allais, RSC Adv., 2013, 3, 8988 RSC.
- F. Pion, P.-H. Ducrot and F. Allais, Macromol. Chem. Phys., 2014, 215, 431 CrossRef CAS.
- M. Z. Oulame, F. Pion, P.-H. Ducrot and F. Allais, Eur. Polym. J., 2015, 63, 186–193 CrossRef CAS.
- I. Barbara, A. L. Flourat and F. Allais, Eur. Polym. J., 2015, 62, 236–243 CrossRef CAS.
- A. F. Reano, J. Cherubin, A. M. M. Peru, Q. Wang, T. Clément, S. Domenek and F. Allais, ACS Sustainable Chem. Eng., 2015, 3, 3486–3496 CrossRef CAS.
- A. F. Reano, F. Pion, S. Domenek, P.-H. Ducrot and F. Allais, Green Chem., 2016 10.1039/c6gc00117c.
- A. S. Jaufurally, A. R. S. Teixeira, L. Hollande, F. Allais, P.-H. Ducrot, unpublished results.
- H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404 RSC.
- O. Kreye, T. Tóth and M. A. R. Meier, Eur. Polym. J., 2011, 47, 1804 CrossRef CAS.
- H. Mutlu and M. A. R. Meier, Macromol. Chem. Phys., 2009, 210, 1019 CrossRef CAS.
- L. M. de Espinosa, M. A. R. Meier, J. C. Ronda, M. Galià and V. Cadiz, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1649 CrossRef.
- T. W. Baughman and K. B. Wagener, Adv. Polym. Sci., 2005, 176, 1 CrossRef CAS.
- O. Türünç, L. M. de Espinosa and M. A. R. Meier, Macromol. Rapid Commun., 2011, 32, 1357 CrossRef PubMed.
- S. Günther, P. Lamprecht and G. A. Luinstra, Macromol. Symp., 2010, 293, 15 CrossRef.
- M. Firdaus and M. A. R. Meier, Eur. Polym. J., 2013, 49, 156 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 7693–7700 RSC.
- T. Lebarbé, A. S. More, P. S. Sane, E. Grau, C. Alfos and H. Cramail, Macromol. Rapid Commun., 2014, 35, 479–483 CrossRef PubMed.
- T. Lebarbé, M. Neqal, E. Grau, C. Alfos and H. Cramail, Green Chem., 2014, 16, 1755–1758 RSC.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Eur. Polym. J., 2015, 67, 409–417 CrossRef CAS.
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 8963 CrossRef CAS.
- M. D. Schulz and K. B. Wagener, ACS Macro Lett., 2012, 1, 449 CrossRef CAS.
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubb, J. Am. Chem. Soc., 2005, 31, 368 Search PubMed.
- M. Abbas and C. Slugovc, Tetrahedron Lett., 2011, 52, 2560 CrossRef CAS.
- H. T. H. Nguyen, M. H. Reis, P. Qi and S. A. Miller, Green Chem., 2015, 17, 4512 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06348a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |