
Carbon nanodots-catalyzed free radical polymerization of water-soluble vinyl monomers†
Abstract
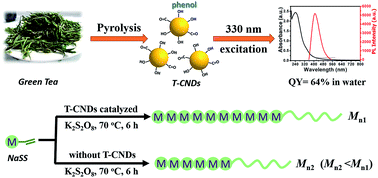
Carbon nanodots made from green tea (T-CNDs) catalyzing free radical polymerization of water-soluble vinyl monomers is reported. T-CNDs-catalyzed polymerization yields a higher molecular weight as well as a narrower polydispersity index than without T-CNDs, which is attributed to good electron accepting ability of T-CNDs and redox initiators formation.
 Please wait while we load your content...
Please wait while we load your content...

