
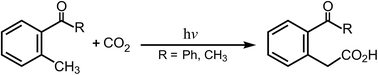
Mechanistic analysis of the photochemical carboxylation of o-alkylphenyl ketones with carbon dioxide†
Abstract
The mechanisms for photochemical carboxylation reactions are studied theoretically using two model systems: o-methylbenzophenone and o-methylacetophenone, with the M06-L and the 6-311G(d,p) basis set. Three reaction routes are used to ascertain the actual photochemical reaction mechanism for the o-alkylphenyl ketone molecules. The structures of the intersystem crossing, which play a central role in these photocarboxylations, are determined. The intermediates and transition structures for the ground singlet states and the excited triplet states are also computed to allow a qualitative interpretation of the entire reaction path. The theoretical findings show that when o-alkylphenyl ketone molecules are produced in the T1 state by photoexcitation at 365 nm, intersystem crossing to the S0 surface is the most probable pathway for deactivation. When they relax to the S0 state, the o-alkylphenyl ketone molecules can either react with CO2 to undergo the insertion reaction, which produces the carboxylic acid, or revert to the reactants. In particular, the theoretical investigations strongly suggest that the insertion of CO2 in these photocarboxylation reactions for o-alkylphenyl ketones occurs on the S0 surface, rather than on the excited T1 surface.
 Please wait while we load your content...
Please wait while we load your content...

