
Aqueous phase hydrogenation of furfural to tetrahydrofurfuryl alcohol on alkaline earth metal modified Ni/Al2O3†
Abstract
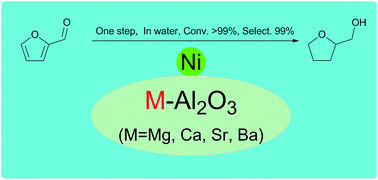
Al2O3 modified by alkaline earth metals M–Al2O3 (M = Mg, Ca, Sr, Ba) was synthesised by coprecipitation method. The nickel-based catalysts supported by M–Al2O3 were prepared by impregnation method. The catalysts were characterized by TEM, N2 adsorption/desorption, XRD, H2-TPR, NH3-TPD and XPS, and used for the direct hydrogenation of furfural to tetrahydrofurfuryl alcohol (THFA) in water. The reaction was demonstrated to proceed through furfuryl alcohol as an intermediate. The modification of Al2O3 by alkaline earth metals has a significant effect on the activity and selectivity of THFA. A high yield of THFA was obtained over Ni/Ba–Al2O3 under optimized conditions. Moreover, the catalyst is recyclable and reusable at least four times without significant loss of the conversion of furfural and selectivity of THFA.
 Please wait while we load your content...
Please wait while we load your content...

