
Biomimetic tumor-induced angiogenesis and anti-angiogenic therapy in a microfluidic model
Abstract
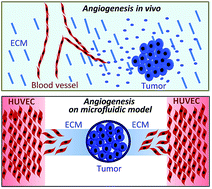
We developed a biomimetic microfluidic model to reproduce hallmark events of tumor-induced angiogenesis. The angiogenic capabilities of salivary gland adenoid cystic carcinoma (ACC) cells and oral squamous cell carcinoma (SCC) cells were assessed in this model. The traditional nude mouse xenograft model was used to investigate the physiological similarity of the microfluidic model to animal models, and the results showed that the angiogenic potential of ACC and SCC cells assessed by the microfluidic model was in agreement with the results obtained from the nude mouse model. The microfluidic model was subsequently used to evaluate the effect of antiangiogenic drugs on ACC- and SCC-induced angiogenesis. The antiangiogenic effect of anti-VEGF was further compared between the microfluidic and nude mouse models, and showed that it effectively inhibited tumor-induced angiogenesis in both the microfluidic model and the nude mouse model. Thus, tumor-induced angiogenesis reproduced in the microfluidic model may expand the capabilities of cell culture models, providing a low-cost, time-saving, and rapid alternative to animal models.
 Please wait while we load your content...
Please wait while we load your content...

