
Racemic alkaloids from Macleaya cordata: structural elucidation, chiral resolution, and cytotoxic, antibacterial activities†
Chun-Mei Sai‡
a,
Da-Hong Li‡a,
Sheng-Ge Lia,
Tong Hana,
Yong-Zhi Guoa,
Yue-Hu Peia,
Jiao Baia,
Yong-Kui Jingb,
Zhan-Lin Li*a and
Hui-Ming Hua*a
aKey Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, P. R. China. E-mail: huimhua@163.com; lzl1030@hotmail.com; Tel: +86-024-23986465
bDepartment of Medicine, Mount Sinai School of Medicine, New York, New York 10029, USA
First published on 20th April 2016
Abstract
Three pairs of new enantiomeric natural alkaloids (±)-macleayins C–E (1–3), together with five pairs of known racemic alkaloids (4–8), were isolated from the aerial parts of Macleaya cordata. Compounds 1–5 were separated successfully by chiral-phase HPLC to yield optically pure isomers. However, chiral resolution of compounds 6–8 existing as racemers in the plant was unsuccessful. It is noteworthy that, macleayin C represents a novel type of hybrid composed of a dihydrobenzophenanthridine alkaloid and a phenylpropanoid. The structures including the absolute configurations of (±)-1–5 were established by detailed spectroscopic analyses and electronic circular dichroism calculations. All the isolates were evaluated for in vitro anti-tumor and antibacterial activities, and compounds (±)-4–5 exhibited potent cytotoxicity against HL-60 cell lines with IC50 values less than 3.0 μM, and compounds (±)-1–3 showed moderate cytotoxic activity. Only compound 7 revealed inhibitory activities against Staphylococcus aureus, Bacillus subtilis, and Candida albicans with MIC values of 33.07, 8.27, and 8.27 μg mL−1, respectively.
Introduction
Macleaya cordata (Willd.) R. Br., a deciduous perennial plant in the family Papaveraceae, is a common traditional Chinese medicine, and has recently attracted much attention due to its bioactive alkaloid contents and pharmaceutical values.1–3 Many studies have demonstrated that benzophenanthridine, dihydrobenzophenanthridine, protopine, and protoberberine type alkaloids are the major bioactive components in the plant.4 Experimental research showed that M. cordata has a wide spectrum of anti-microbial,5 anti-fungal,6 anti-inflammatory,7 pesticidal,8 and anti-tumor properties.9,10 According to clinical records, M. cordata has been applied extensively as folk medicine in China, North America, and Europe, where it has been used to cure cervical cancer and thyroid cancer,3 and as traditional medicines for the treatment of diverse infectious diseases.11 In addition, it was also successfully applied in veterinary medicine and agriculture. Preliminary phytochemical investigation on the ethanol extract of M. cordata afforded two pairs of novel enantiomers of benzophenanthridine–protopine hybrids,12 which intrigued us to continue to explore the chemical diversity of the alkaloids, leading to the identification of three pairs of new enantiomers (±)-1–3, along with two pairs of known enantiomers (±)-4–5 and three racemic compounds 6–8 (Fig. 1). Herein we report the isolation, chiral resolution, structural identification, and in vitro cytotoxic and antibacterial activities of the isolated compounds.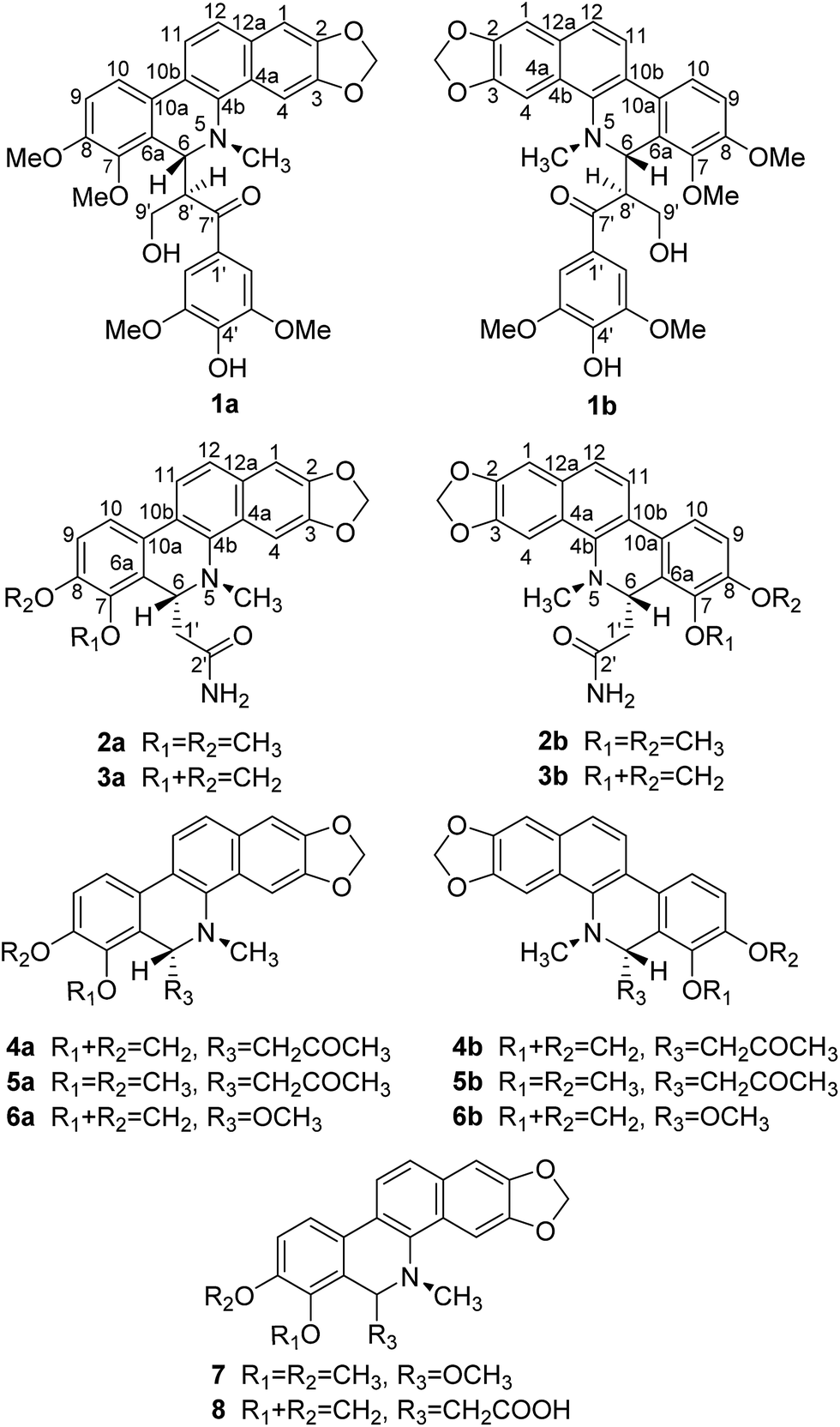 | ||
| Fig. 1 Structures of compounds 1–8. | ||
Results and discussion
All isolated compounds 1–8 displayed strong fluorescence under UV (254 nm and 365 nm) on silica gel TLC plates, as well as positive reactions with Dragendorff's reagent. Their basic skeleton was in perfect accordance with dihydrobenzophenanthridines13 according to the following features: their UV absorptions at about max. 225–230, 280–285, and 318–325 nm, their IR aromatic ring absorption bands at about 1600, 1494, and 1464 cm−1, their 1H NMR spectra with the signals of two one-proton singlets at δH 7.11–7.42 (H-1) and 6.44–7.72 (H-4), two pairs of AB-type ortho-coupling doublets at δH 6.87–7.14 (d, J = 8.1–8.6 Hz, H-9) and 7.34–7.66 (d, J = 8.1–8.6 Hz, H-10), δH 7.70–7.85 (d, J = 8.6–8.7 Hz, H-11) and 7.48–7.56 (d, J = 8.6–8.7 Hz, H-12), as well as one N-methyl group at δH 2.33–2.79 (s). All of the 1H NMR spectra also exhibited a typical signal of H-6 at δH 4.77–5.55, which was from a dihydrobenzophenanthridine skeleton substituted at C-6 by a directly connected methine.Macleayin C (1) was obtained as a colourless cluster crystal (in CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 1:1), [α]25D 0 (c 0.16, CHCl3). The molecular formula of C32H31NO9 was determined by the positive HRESIMS ion at m/z 596.1892 [M + Na]+ (calcd 596.1891), implying eighteen indices of hydrogen deficiency. The UV spectrum showed the maximum absorptions at 227, 281, and 320 nm. The IR spectrum indicated the presence of a carbonyl (1659 cm−1) and aromatic ring (1605, 1492, and 1464 cm−1). The 1H and 13C NMR spectra (Table 1) of 1 revealed a typical 6-substituted-dihydrochelerythrine moiety12 with signals at δH 7.14 (1H, d, J = 8.6 Hz, H-9), 7.66 (1H, d, J = 8.6 Hz, H-10), 7.85 (1H, d, J = 8.6 Hz, H-11), 7.53 (1H, d, J = 8.6 Hz, H-12), 7.12 (1H, s, H-1), 6.44 (1H, s, H-4), 4.57 (1H, d, J = 11.0 Hz, H-6), 6.07, 5.81 (each d, J = 1.1 Hz, 2,3-OCH2O), 3.84 (3H, s, 7-OCH3), 3.90 (3H, s, 8-OCH3), 2.33 (3H, s, N–CH3); δC 103.3 (C-1), 146.8 (C-2), 146.8 (C-3), 99.6 (C-4), 126.5 (C-4a), 138.9 (C-4b), 58.4 (C-6), 125.9 (C-6a), 145.9 (C-7), 151.7 (C-8), 112.3 (C-9), 119.2 (C-10), 124.2 (C-10a), 123.0 (C-10b), 119.4 (C-11), 123.6 (C-12), 130.1 (C-12a), 100.9 (2,3-OCH2O), 60.5 (7-OCH3), 55.8 (8-OCH3), 41.6 (N–CH3). The above deduction was confirmed by the key HMBC correlations from H-1 to C-3, C-4a, and C-12, H-4 to C-2, C-4b, and C-12a, H-9 to C-7 and C-10a, H-10 to C-6a and C-10b, H-11 to C-4b, C-10a, and C-12a, H-12 to C-1, C-4a, and C-10b, N–CH3 to C-4b and C-6, as shown in Fig. 2.
:MeOH = 1:1), [α]25D 0 (c 0.16, CHCl3). The molecular formula of C32H31NO9 was determined by the positive HRESIMS ion at m/z 596.1892 [M + Na]+ (calcd 596.1891), implying eighteen indices of hydrogen deficiency. The UV spectrum showed the maximum absorptions at 227, 281, and 320 nm. The IR spectrum indicated the presence of a carbonyl (1659 cm−1) and aromatic ring (1605, 1492, and 1464 cm−1). The 1H and 13C NMR spectra (Table 1) of 1 revealed a typical 6-substituted-dihydrochelerythrine moiety12 with signals at δH 7.14 (1H, d, J = 8.6 Hz, H-9), 7.66 (1H, d, J = 8.6 Hz, H-10), 7.85 (1H, d, J = 8.6 Hz, H-11), 7.53 (1H, d, J = 8.6 Hz, H-12), 7.12 (1H, s, H-1), 6.44 (1H, s, H-4), 4.57 (1H, d, J = 11.0 Hz, H-6), 6.07, 5.81 (each d, J = 1.1 Hz, 2,3-OCH2O), 3.84 (3H, s, 7-OCH3), 3.90 (3H, s, 8-OCH3), 2.33 (3H, s, N–CH3); δC 103.3 (C-1), 146.8 (C-2), 146.8 (C-3), 99.6 (C-4), 126.5 (C-4a), 138.9 (C-4b), 58.4 (C-6), 125.9 (C-6a), 145.9 (C-7), 151.7 (C-8), 112.3 (C-9), 119.2 (C-10), 124.2 (C-10a), 123.0 (C-10b), 119.4 (C-11), 123.6 (C-12), 130.1 (C-12a), 100.9 (2,3-OCH2O), 60.5 (7-OCH3), 55.8 (8-OCH3), 41.6 (N–CH3). The above deduction was confirmed by the key HMBC correlations from H-1 to C-3, C-4a, and C-12, H-4 to C-2, C-4b, and C-12a, H-9 to C-7 and C-10a, H-10 to C-6a and C-10b, H-11 to C-4b, C-10a, and C-12a, H-12 to C-1, C-4a, and C-10b, N–CH3 to C-4b and C-6, as shown in Fig. 2.
| Position | 13C NMR | 1H NMR (J in Hz) | HMBC (H → C) |
|---|---|---|---|
| a 1H NMR recorded at 600 MHz, 13C NMR recorded at 100 MHz. | |||
| 1 | 103.3 | 7.12 (s) | 3, 4a, 12 |
| 2 | 146.8 | ||
| 3 | 146.8 | ||
| 4 | 99.6 | 6.44 (s) | 2, 4b, 12a |
| 4a | 126.5 | ||
| 4b | 138.9 | ||
| 6 | 58.4 | 4.57 (d, 11.0) | 4b, 7, 10a, 7′, 8′, 9′, N–CH3 |
| 6a | 125.9 | ||
| 7 | 145.9 | ||
| 8 | 151.7 | ||
| 9 | 112.3 | 7.14 (d, 8.6) | 7, 8, 10a |
| 10 | 119.2 | 7.66 (d, 8.6) | 6a, 8, 10b |
| 10a | 124.2 | ||
| 10b | 123.0 | ||
| 11 | 119.4 | 7.85 (d, 8.6) | 4b, 10a, 12a |
| 12 | 123.6 | 7.53 (d, 8.6) | 1, 4a, 10b |
| 12a | 130.1 | ||
| 1′ | 128.8 | ||
| 2′,6′ | 105.5 | 6.58 (s) | 4′, 7′ |
| 3′,5′ | 146.7 | ||
| 4′ | 140.3 | ||
| 7′ | 201.0 | ||
| 8′ | 49.5 | 3.54 (ddd, 11.0, 10.6, 3.6) | 6, 6a, 7′, 9′ |
| 9′ | 61.8 | 3.82 (ddd, 10.6, 10.4, 7.4) | 7′, 8′ |
| 3.16 (ddd, 10.4, 3.7, 3.6) | |||
| OCH2O | 100.9 | 5.81, 6.07 (each d, 1.1) | 2, 3 |
| N–CH3 | 41.6 | 2.33 (s) | 4b, 6 |
| 7-OCH3 | 60.5 | 3.84 (s) | 7 |
| 8-OCH3 | 55.8 | 3.90 (s) | 8 |
| 3′,5′-OCH3 | 55.3 | 3.41 (s) | 3′, 5′ |
| 4′-OH | 8.96 (br s) | ||
| 9′-OH | 4.21 (dd, 7.4, 3.7) | 8′, 9′ | |
 | ||
| Fig. 2 Selected 2D NMR correlations of 1–3. | ||
The remaining data in the 1H and 13C NMR spectra exhibited signals for a 1,3,4,5-tetrasubstituted benzene ring [δH 6.58 (2H, s, H-2′,6′), 8.96 (1H, br s, 4′-OH), 3.41 (6H, s, 3′,5′-OCH3); δC 128.8 (C-1′), 105.5 (C-2′,6′), 146.7 (C-3′,5′), 140.3 (C-4′), 55.3 (3′,5′-OCH3)], a ketone group (δC 201.0, C-7′), a hydroxyethyl side chain [δH 3.54 (1H, ddd, J = 11.0, 10.6, 3.6 Hz, H-8′), 3.82 (1H, ddd, J = 10.6, 10.4, 7.4 Hz, H-9′a), 3.16 (1H, ddd, J = 10.4, 3.7, 3.6 Hz, H-9′b), 4.21 (1H, dd, J = 7.4, 3.7 Hz, 9′-OH); δC 49.5 (C-8′), 61.8 (C-9′)]. The above data suggested the presence of 3,5-dimethoxy-4-hydroxyphenyl 2-hydroxyethyl ketone moiety, which was confirmed by the 1H–1H COSY correlations of OH-9′/H-9′/H-8′ and the HMBC correlations from OH-9′ to C-8′ and C-9′, and from H-8′ and H-2′/6′ to C-7′. The 1H–1H COSY correlation of H-6/H-8′ and the HMBC correlations from H-6 to C-7′, C-9′, C-4b, C-10a, and N–CH3 further established the linkage of dihydrochelerythrine and phenyl ethyl ketone via C-6 and C-8′. By detailed analyses of the 1D and 2D NMR data, the planar structure was deduced to be shown in 1. The relative configuration of macleayin C (1) was established by the coupling constant of H-6 and H-8 (11.0 Hz), and analyses of the NOESY correlations (Fig. 3) from H-6 to N–CH3 and H-9′b, from H-8′ to H-9′a indicated that H-6 and H-8′ were located on the opposite side.
 | ||
| Fig. 3 NOESY correlations of 1. | ||
The isolated sample of 1 was a racemic, due to its specific optical rotation of [α]25D 0 (c 0.16, CHCl3). Subsequent separation by using chiral-phase HPLC yielded (−)-macleayin C (1a) (1.80 mg) and (+)-macleayin C (1b) (1.80 mg) in a ratio of 1:1 (Fig. 4), whose optical rotations were opposite.13 Furthermore, they have the similar absolute value of specific rotation, [α]20D = −85.6 (c 0.18, MeOH) and +94.4 (c 0.18, MeOH), respectively. The assignments of 1a (6R, 8′R) and 1b (6S, 8′S) were made by comparing the calculated electronic circular dichroisms (ECD) via a quantum method with the experimental data (Fig. 5).14 From the above evidence, the structures of 1a and 1b were unambiguously assigned as shown in Fig. 1.
 | ||
| Fig. 4 The chiral HPLC chromatograms of 1–8. | ||
 | ||
| Fig. 5 Experimental and suitable calculated ECD spectra of (±)-1. | ||
Macleayin D (2) was isolated as a colourless cluster crystal (in MeOH) with [α]25D 0 (c 0.20, CHCl3). HRESIMS (m/z 407.1602 [M + H]+, calcd 407.1601) and 13C NMR data established the molecular formula C23H22N2O5. The UV absorption bands at 229, 283, and 318 nm indicated typical conjugated groups. The IR spectrum showed absorption bands due to amino (3382 cm−1), carbonyl (1680 cm−1), and aromatic ring (1602, 1494, and 1465 cm−1) functionalities. The comparison of 1H and 13C NMR data of 2 with 1 clearly showed that they have the same 6-substituted dihydrochelerythrine moiety (Tables 1 and 2). The remaining signals of 2 in the 1H and 13C NMR as well as HSQC spectra established the presence of an acetamido group at δH 6.98 (1H, br s, –NH2), 6.74 (1H, br s, –NH2), 2.06 (1H, dd, J = 14.7, 11.3 Hz, H-1′a), 1.90 (1H, dd, J = 14.7, 3.5 Hz, H-1′b); δC 39.6 (C-1′), 171.5 (C-2′). The HMBC correlations (Fig. 2) of –NH2/C-1′, C-2′ and H-1′/C-6, C-2′ further confirmed the presence of acetamido group, which was assigned to be substituted at C-6. Thus, compound 2 was elucidated as 6-acetamidyldihydrochelerythrine. Subsequent chiral resolution of 2 was performed on a chiral column (Daicel Chiralpak IC) to yield 2a (0.97 mg) and 2b (0.97 mg) in ratio of 1:1 (Fig. 4), of which they were virtually opposite in terms of their ECD curves and optical rotation data [α]20D (c 0.097, MeOH) −126.3 (2a), [α]20D (c 0.097, MeOH) +142.8 (2b). Their absolute configurations were determined by the quantum chemical ECD calculation method. The measured ECD curves of 2a and 2b matched with the calculated ones of (6R)-2 and (6S)-2, respectively (Fig. 6). Thus, the absolute configurations of them were established as 2a (6R) and 2b (6S) as shown in Fig. 1.
| Position | 2 | 3 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| a 1H NMR recorded at 600 MHz, 13C NMR recorded at 150 MHz. | ||||
| 1 | 104.1 | 7.29 (s) | 104.1 | 7.29 (s) |
| 2 | 147.2 | 147.3 | ||
| 3 | 147.6 | 147.7 | ||
| 4 | 100.1 | 7.45 (s) | 100.1 | 7.47 (s) |
| 4a | 126.8 | 127.0 | ||
| 4b | 139.2 | 139.1 | ||
| 6 | 54.1 | 4.92 (dd, 11.2, 3.4) | 54.0 | 4.77 (dd, 11.0, 3.4) |
| 6a | 128.2 | 116.2 | ||
| 7 | 145.0 | 144.0 | ||
| 8 | 151.9 | 146.9 | ||
| 9 | 111.8 | 7.08 (d, 8.6) | 107.4 | 6.95 (d, 8.2) |
| 10 | 118.7 | 7.64 (d, 8.6) | 116.3 | 7.45 (d, 8.2) |
| 10a | 124.0 | 125.0 | ||
| 10b | 122.8 | 122.8 | ||
| 11 | 119.8 | 7.81 (d, 8.6) | 120.0 | 7.80 (d, 8.6) |
| 12 | 123.5 | 7.53 (d, 8.6) | 123.7 | 7.54 (d, 8.6) |
| 12a | 130.7 | 130.7 | ||
| 1′ | 39.6 | 2.06 (dd, 14.7, 11.3) | 39.6 | 2.13 (dd, 14.7, 11.1) |
| 1.90 (dd, 14.7, 3.5) | 1.97 (dd, 14.5, 2.8) | |||
| 2′ | 171.5 | 171.3 | ||
| NH2 | 6.98, 6.74 (each br s) | 7.03, 6.77 (each br s) | ||
| N–CH3 | 42.7 | 2.53 (s) | 43.1 | 2.54 (s) |
| 2,3-OCH2O | 101.1 | 6.13, 6.12 (each br s) | 101.2 | 6.14, 6.13 (each br s) |
| 7-OCH3 | 60.4 | 3.84 (s) | ||
| 8-OCH3 | 55.7 | 3.87 (s) | ||
| 7,8-OCH2O | 101.5 | 6.11 (br s) | ||
 | ||
| Fig. 6 Experimental and suitable calculated ECD spectra of (±)-2. | ||
Macleayin E (3) was obtained as a colourless cluster crystal (in MeOH) and displayed an [M + Na]+ ion peak at m/z 413.1110 (calcd 413.1108), corresponding to a molecular formula of C22H18N2O5. Its IR and UV spectra were similar to those of compound 2. Based on a comparison of the NMR spectroscopic data of 3 with 2 (Table 2), they had the same dihydrobenzophenanthridine skeleton and the same 6-acetamido side chain, except for the appearance of signals for one more methylenedioxy group and the absence of two methoxyl groups in 3. The additional methylenedioxy group was determined to be attached to C-7 and C-8 by HMBC correlations from the protons of the methylenedioxy [δH 6.11 (2H, s)] to C-7 (δC 144.0) and C-8 (δC 146.9). Accordingly, compound 3 was established as 6-acetamidyldihydrosanguinarine, which was isolated in a racemic form. The chiral HPLC separation of 3 was performed by the same method with 2 to yield 3a (0.95 mg) and 3b (0.95 mg). Subsequently, the assignment of 3a (6R) ([α]20D (c 0.095, MeOH) −143.8), and 3b (6S) ([α]20D (c 0.095, MeOH) +138.1) was accomplished by comparison of the experimental ECD spectra with those of 2a and 2b (Fig. 7).
 | ||
| Fig. 7 Experimental ECD spectra of (±)-2, 3, 4, and 5. | ||
6-Acetonyldihydrosanguinarine (4) and 6-acetonyldihydrochelerythrine (5) were also isolated as known compounds. Their structures were identified by comparison of their 1H and 13C NMR data with literature values.15 They were also in racemic form in M. cordata, and then subjected to chiral column chromatography respectively to obtain 4a (1.80 mg), 4b (1.80 mg), 5a (0.40 mg), and 5b (0.40 mg) (Fig. 4). Their experimental ECD spectra were compatible with those of 2a and 2b (Fig. 7), respectively, establishing their absolute configurations as 4a (6R), 4b (6S), 5a (6R), and 5b (6S). This is the first report of their chiral resolution (Table 3).
| Compound | IC50 (μM) | ||
|---|---|---|---|
| HL-60 | A-549 | MCF-7 | |
| (+)-1 | 10.35 | >100 | >100 |
| (−)-1 | 12.13 | >100 | >100 |
| (+)-2 | 25.42 | 34.19 | >100 |
| (−)-2 | 26.17 | 35.26 | >100 |
| (+)-3 | 20.97 | 30.46 | >100 |
| (−)-3 | 18.46 | 31.57 | >100 |
| (+)-4 | 2.07 | 22.47 | 42.74 |
| (−)-4 | 1.86 | 27.14 | 45.63 |
| (+)-5 | 2.75 | 25.45 | 49.63 |
| (−)-5 | 2.58 | 22.14 | 48.06 |
| 5-Fu | 2.80 | 1.60 | 29.83 |
The known compounds 6-methoxyldihydrosanguinarine (6),16 6-methoxyldihydrochelerythrine (7),17,18 and spallidamine (8)19,20 were obtained. According to related literature and NMR data, their structures were determined. Due to their ECD curves almost in a straight line and the lack of optical activity, they existed as racemic natures. Subsequently, enantiomers of 6 were separated by chiral HPLC, while the isomers rapidly formed a racemic mixture. This unusually rapid racemization may originate from the formation of a stable iminium ion intermediate, sanguinarine.21,22 Unfortunately, compounds 7 and 8 could not be separated using three types of chiral columns.
Considering that some benzophenanthridine alkaloids from M. cordata have been reported showing significant anti-tumor, antimicrobial, and antifungal activities, compounds (±)-1–5 were tested for cytotoxicity against three human cancer cell lines HL-60, A-549, and MCF-7, and all compounds were tested for antimicrobial activity against Escherichia coli, Staphylococcus aureus, Bacillus subtilis, and Candida albicans.
Conclusions
In summary, three pairs of new enantiomers (±)-macleayins C-E (1–3), together with two pairs of known enantiomers (±)-4–5, and three racemic alkaloids (6–8) were isolated from Macleaya cordata, and the structures were elucidated by extensive spectroscopic techniques. All isolated 6-substituted dihydrobenzophenanthridine alkaloids from M. cordata are raceme. Compounds 1–5 were separated by using chiral-phase HPLC. Their absolute configurations were determined by calculated ECD method.The cytotoxicity of compounds (±)-1–5 against three human cancer cell lines was evaluated using trypan blue and MTT methods. Compounds (±)-4 and 5 exhibited potent cytotoxicity against HL-60 cell lines, and (±)-1–3 showed moderate cytotoxicity. However, compounds (±)-1–5 showed weak or no cytotoxic activities against A-549 and MCF-7 cell lines. The results indicated that (+)-1–5 and (−)-1–5 had almost the same cytotoxicity, scilicet no selectivity, perhaps because that the enantiomer was liable to undergo racemization during incubating.
Compounds 1–8 were tested for antimicrobial activities by micro-dilution broth MIC method. However, only compound 7 showed mild effects.
Experimental section
General experimental procedures
Optical rotations were measured with Rudolph Autopol-V digital polarimeter and Perkin Elmer Model 341 Polarimeter. UV spectra were recorded on a Shimadzu UV-2201 spectrometer. IR spectra were obtained on a Bruker IFS-55 spectrometer using KBr disks. ECD spectra were acquired with a Bio-logic MOS 450 spectropolarimeter. 1D and 2D NMR were performed on Bruker ARX-300 and AV-600 NMR spectrometers using solvent signals (DMSO-d6: δH 2.50/δC 39.52; CDCl3: δH 7.26/δC 77.16), with tetramethylsilane (TMS) as the internal standard. HRESIMS spectra were collected using a Bruker micrOTOF-Q mass spectrometer. A Shimadzu LC-6 AD equipped with a SPD-20A (UV/VIS) detector was used for HPLC, a YMC-pack ODS-A column (250 × 20 mm, S-5 μm, 12 nm) was used for semi-preparative HPLC separation. A Shimadzu LC-20AB equipped with a SPD-M20A PDA (DIODE ARRAY) detector on a chiral column Daicel Chiralpak IC (250 × 4.6 mm) was used for chiral HPLC separation. Column chromatography (CC) were performed with silica gel (100–200 and 200–300 mesh, Qingdao Haiyang Chemical Co. Ltd., Qingdao, China), C18 reversed-phase silica gel (S-50 μm, 12 nm, YMC Co. Ltd., Kyoto, Japan), and Sephadex LH-20 gel (GE Healthcare, Sweden). Fractions obtained from CC were monitored by TLC using precoated silica gel GF254 plates (Qingdao Haiyang Chemical Co. Ltd., Qingdao, China), which was visualized under UV light, by spraying with Dragendorff's reagent, or heating the silica gel plates after being sprayed with 10% H2SO4 in EtOH.Plant material
The aerial parts of Macleaya cordata (Willd.) R. Br. was purchased from Anguo Medicines Ltd. (Hebei, China) in November 2013, and was identified by Prof. Jin-Cai Lu (School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang, China). A voucher specimen (BLH-20131108) has been deposited at the Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang, China.Extraction and isolation
The air-dried aerial parts of M. cordata (40.0 kg) were extracted with 95% EtOH (2 × 400 L) under reflux, and 75% EtOH (1 × 400 L). After the organic solvent was removed under reduced pressure, the crude extract was suspended in H2O, successively partitioned with CH2Cl2 and n-BuOH. The dichloromethane extract (500 g) was fractionated on a silica gel column (200–300 mesh; 1000 g; Φ 10 cm × 120 cm) and eluted with petroleum ether (60–90 °C)–acetone (100:5, 100:10, 100:20, 100:50, 100:100 and 0:100, v/v) to yield six fractions (A–F). Fraction D (50 g) was subjected to Rp-C18 CC (50 μm; 180 g; Φ 6 cm × 45 cm) eluting with MeOH–H2O (50:50, 60:40, 65:35, 70:30, 80:20, 90:10 and 100:0, v/v) to afford seven major subfractions (D1–D7). Subfraction D2 was separated by reversed-phase preparative HPLC (MeOH–H2O, 75:25, v/v; flow rate, 5 mL min−1) to give four fractions (D2-1–D2-4). D2-2 was further purified by preparative HPLC procedure (MeOH–H2O, 70:30, v/v; flow rate, 5 mL min−1) to yield compounds 1 (8 mg, tR = 32.6 min), 2 (6 mg, tR = 84.5 min), and 3 (6 mg, tR = 90.2 min). Subfraction D3 was separated by reversed-phase preparative HPLC (MeOH–H2O, 75:25, v/v; flow rate, 5 mL min−1), followed by purification using Sephadex LH-20 with CH2Cl2–MeOH (1:1) to afford 4 (15 mg) and 5 (16 mg). Fraction E (70 g) was loaded onto a silica gel column (200–300 mesh; 650 g; Φ 5.3 cm × 140 cm) using CH2Cl2–MeOH (100:0, 100:1, 100:2, 100:3, 100:5, and 0:100, v/v) as the eluting reagent to furnish six subfractions (E1–E6). Subfraction E1 (8 g) was chromatographed over Rp-C18 CC (50 μm; 150 g; Φ 4 cm × 50 cm) eluted with MeOH–H2O (30:70, 45:55, 50:50, 55:45, 65:35, 75:25, 0:100, v/v) to give seven fractions (E1a–E1g). Fraction E1b was purified by Sephadex LH-20 with CH2Cl2–MeOH (1:1) to afford compound 8 (4 mg). Fraction E1e was purified by Sephadex LH-20 with CH2Cl2–MeOH (1:1) to yield 6 (5 mg) and 7 (5 mg). The chiral HPLC separations of 1–5 were performed on a chiral column (Daicel Chiralpak IC) to yield 1a (1.80 mg), 1b (1.80 mg), 2a (0.97 mg), 2b (0.97 mg), 3a (0.95 mg), 3b (0.95 mg), 4a (1.80 mg), 4b (1.80 mg), 5a (0.40 mg), and 5b (0.40 mg), respectively.
Characterization of new compounds
ε) 227 (4.6), 281 (4.6), 320 (4.0) nm; IR (KBr) νmax 3470, 2926, 1659, 1605, 1492, 1464 cm−1; 1H and 13C NMR data, see Table 1; positive HRESIMS m/z 596.1892 [M + Na]+ (calcd for C32H31NO9Na, 596.1891). (−)-1a: [α]20D = −85.6 (c 0.18, MeOH), (+)-1b: [α]20D = +94.4 (c 0.18, MeOH).ε) 229 (4.6), 283 (4.6), 318 (4.2) nm; IR (KBr) νmax 3382, 2926, 2854, 2798, 1680, 1602, 1494, 1465 cm−1; 1H and 13C NMR data, see Table 2; positive HRESIMS m/z 407.1602 [M + H]+ (calcd for C23H23N2O5, 407.1601). (−)-2a: [α]20D = −126.3 (c 0.097, MeOH), (+)-2b: [α]20D = +142.8 (c 0.097, MeOH).ε) 236 (4.6), 286 (4.6), 323 (4.2) nm; IR (KBr) νmax 3397, 2895, 1675, 1602, 1496, 1464 cm−1; 1H and 13C NMR data, see Table 2; positive HRESIMS m/z 413.1110 [M + Na]+ (calcd for C22H18N2O5Na, 413.1108). (−)-3a: [α]20D = −143.8 (c 0.095, MeOH), (+)-3b: [α]20D = +138.1 (c 0.095, MeOH).Biological assays
In the trypan blue method, cells in logarithmic growth were seeded at 5 × 104 cells per mL in 24-well microplates and incubated with various concentrations of the compounds under a humidified atmosphere of 5% CO2 and 95% air at 37 °C for 3 days. The compounds were dissolved in DMSO and then diluted to the proper concentrations. Cell viability was determined after staining the cells with trypan blue. Trypan blue-stained (nonviable) cells and the total cell number were determined with a hemocytometer. The growth inhibition in cells after treatment with different concentrations was calculated comparing with control cells (5-fluorouracil was used as a positive control), and a half growth inhibitory concentration (IC50) was obtained by regression analysis of the concentration response data.24
In the MTT assay, briefly, cells suspensions, 200 μL, at a density of 2.5 × 104 cells per mL, were seeded into 96-well microtiter plates and incubated for 24 h at 37 °C under 5% CO2 and 95% air. Then the tested compounds with different concentrations in DMSO were placed into each microtiter plates and further incubated for 72 h. Finally, 50 μL 0.4% MTT solution was added to each well and incubated for 4 h. Then, the MTT was removed from the wells and the fromazan crystals were dissolved in DMSO (200 μL) for 10 min with shaking. Then the plate was read immediately on a microtiter plate reader (Bio-RAD) at a wavelength of 570 nm to record the optical density (OD). The IC50 value was defined as the concentration of the control in the MTT assay. 5-Fluorouracil (5-Fu) was used as a positive control. All the IC50 results were expressed as average of three independent experiments.25
:1) in different concentrations (0.2–500 μg mL−1) and added in broth medium (bacteria)/Sabouraud (fungus) with inocula [preliminary analyses with DMSO/hydrogenated castor oil (3:1) do not inhibit the growth of the test organisms]. The microplates (bacteria) were incubated at 37 °C for 18–24 h, and the microplates (fungus) at 28 °C for 24–28 h. At the lowest concentration, the microorganisms did not show growth as judged by the presence of turbidity.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (Grant No. 81172958), the Basic Research Subject of Key Laboratory Supported by Educational Commission of Liaoning Province of China (No. LZ2014044). We gratefully acknowledge Mr Yi Sha and Mrs Wen Li, Department of Analytical Testing Center, Shenyang Pharmaceutical University, for measurement of the NMR data.Notes and references
- M. Liu, Y. L. Lin, X. R. Chen, C. C. Liao and W. K. Poo, Exp. Toxicol. Pathol., 2013, 65, 775–787 CrossRef PubMed.
- M. Zhong, K. L. Huang, J. G. Zeng, S. Li, J. M. She, G. Y. Li and L. Zhang, J. Sep. Sci., 2010, 33, 2160–2167 CrossRef CAS PubMed.
- G. P. Pi, P. Ren, J. M. Yu, R. F. Shi, Z. Yuan and C. H. Wang, J. Chromatogr. A, 2008, 1192, 17–24 CrossRef CAS PubMed.
- Z. X. Qing, P. Cheng, X. B. Liu, Y. S. Liu, J. G. Zeng and W. Wang, Rapid Commun. Mass Spectrom., 2014, 28, 1033–1044 CrossRef CAS PubMed.
- P. Kosina, J. Gregorova, J. Gruz, J. Vacek, M. Kolar, M. Vogel, W. Roos, K. Naumann, V. Simanek and J. Ulrichova, Fitoterapia, 2010, 81, 1006–1012 CrossRef CAS PubMed.
- X. J. Wang, C. L. Min, M. Ge and R. H. Zuo, Curr. Microbiol., 2014, 68, 336–341 CrossRef CAS PubMed.
- G. Y. Zuo, F. Y. Meng, J. Han, X. Y. Hao, G. C. Wang, Y. L. Zhang and Q. Zhang, Molecules, 2011, 16, 5453–5459 CrossRef CAS PubMed.
- M. Y. Baek, H. J. Park, G. M. Kim, D. Y. Lee, G. Y. Lee, S. J. Moon, E. M. Ahn, G. S. Kim, M. H. Bang and N. I. Baek, J. Korean Soc. Appl. Biol. Chem., 2013, 56, 135–140 CrossRef CAS.
- J. Ulrichová, Z. Dvořák, J. Vičar, J. Lata, J. Smržová, A. Šedo and V. Šimánek, Toxicol. Lett., 2001, 125, 125–132 CrossRef.
- N. Ahmad, S. Gupta, M. M. Husain, K. M. Heiskanen and H. Mukhtar, Clin. Cancer Res., 2000, 6, 1524–1528 CAS.
- X. L. Yu, X. L. Gao, Z. X. Zhu, Y. Cao, Q. Zhang, P. F. Tu and X. Y. Chai, Molecules, 2014, 19, 13042–13060 CrossRef PubMed.
- C. M. Sai, D. H. Li, C. M. Xue, K. B. Wang, P. Hu, Y. H. Pei, J. Bai, Y. K. Jing, Z. L. Li and H. M. Hua, Org. Lett., 2015, 17, 4102–4105 CrossRef CAS PubMed.
- A. J. Deng and H. L. Qin, Phytochemistry, 2010, 71, 816–822 CrossRef CAS PubMed.
- H. Xiong, G. K. Xiao, G. D. Chen, H. R. Chen, D. Hu, X. X. Li, S. W. Zhong, L. D. Guo, X. S. Yao and H. Gao, RSC Adv., 2014, 4, 24295 RSC.
- F. Feng, Y. F. Zhi, L. C. Lian, L. W. Yuan and X. Ning, Chin. J. Nat. Med., 2012, 10, 0378–0382 CAS.
- G. L. Zhang, G. Rucker, E. Breitmaier and R. Maryer, Phytochemistry, 1995, 40, 1813–1816 CrossRef CAS.
- M. Y. Baek, H. J. Park, G. M. Kim, D. Y. Lee, G. Y. Lee, S. J. Moon, E. M. Ahn, G. S. Kim, M. H. Bang and N. I. Baek, J. Korean Soc. Appl. Biol. Chem., 2013, 56, 135–140 CAS.
- F. Miao, X. J. Yang, L. Zhou, H. J. Hu, F. Zheng, X. D. Ding, D. M. Sun, C. D. Zhou and W. Sun, Nat. Prod. Res., 2011, 25, 863–875 CrossRef CAS PubMed.
- C. Ito, T. Mizuno, T. S. Wu and H. Furukawa, Phytochemistry, 1990, 29, 2044–2045 CrossRef CAS.
- J. F. Zheng, M. J. Qin, Y. Zheng and C. L. Chen, J. China Pharm. Univ., 2007, 38, 112–114 CAS.
- E. Q. Wu, S. H. Jung, J. B. Chen, Y. H. Kim, K. L. Park, W. Lee and J. S. Kang, J. Pharm. Biomed. Anal., 2010, 51, 103–106 CrossRef CAS PubMed.
- C. V. Men, V. K. Sharma, J. B. Chen, H. M. Zhu, E. Q. Wu, W. Lee, Y. S. Jang, Y. H. Kim and S. H. Jung, J. Pharm. Biomed. Anal., 2011, 56, 479–483 CrossRef PubMed.
- K. B. Wang, C. M. Yuan, C. M. Xue, D. H. Li, Y. K. Jing, H. P. He, X. J. Hao, Y. T. Di, Z. L. Li and H. M. Hua, RSC Adv., 2014, 4, 53725–53729 RSC.
- F. Wang, H. M. Hua, Y. H. Pei, D. Chen and Y. K. Jing, J. Nat. Prod., 2006, 69, 807–810 CrossRef CAS PubMed.
- J. Hu, X. D. Shi, J. G. Chen, X. Mao, L. Zhu, L. Yu and J. Y. Shi, Food Chem., 2014, 148, 437–444 CrossRef CAS PubMed.
- J. J. Chen, D. Q. Fei, S. G. Chen and K. Gao, J. Nat. Prod., 2008, 71, 547–550 CrossRef CAS PubMed.
- D. Y. Li, J. X. Wei, H. M. Hua and Z. L. Li, J. Asian Nat. Prod. Res., 2014, 16, 1018–1023 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR, HRESIMS, UV, IR, CD, and quantum chemical ECD calculations of 1–6. See DOI: 10.1039/c6ra05423d |
| ‡ Chun-Mei Sai and Da-Hong Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |