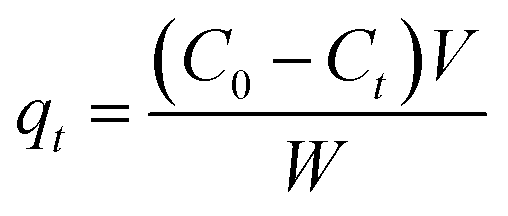DOI:
10.1039/C6RA05253C
(Paper)
RSC Adv., 2016,
6, 50128-50137
Effect of calcination on adsorption performance of Mg–Al layered double hydroxide prepared by a water-in-oil microemulsion method
Received
28th February 2016
, Accepted 14th May 2016
First published on 17th May 2016
Abstract
Herein, Mg–Al layered double hydroxide (LDH) was firstly prepared by a water-in-oil microemulsion method and the prepared LDH sample was further calcined at different temperatures. The calcined LDH samples were carefully characterized using XRD, nitrogen adsorption, TEM, TGA, XPS, FTIR and zeta potential measurements. Calcined and uncalcined LDH samples were used as adsorbents to remove orange II (O-II) dye in water. Adsorption experiments indicated calcination temperatures had an obvious influence on the adsorption affinity of LDH, and the 500 °C calcined LDH sample (LDH-500) exhibited the maximum adsorption capacity of 602 mg g−1 larger than that of LDH (224 mg g−1). The pseudo-second-order model was the best kinetic model to describe O-II adsorbed on the surface of the adsorbent, and the dye adsorption was fitted well using the Langmuir model for the prepared samples. The adsorption mechanism of O-II dye onto the calcined and uncalcined LDHs samples was mainly related to electrostatic interaction between the anionic O-II molecules and positively charged surface of the adsorbents, and the slow intercalation of O-II into the layers of LDH and reconstruction of LDH-500.
Introduction
Synthetic dyes have massively been applied in many industrial processes including dyeing of paper, textiles, leather and plastics. Dye pollutants in groundwater released from these industries can pose serious environmental and public health problems.1,2 A variety of methods, for example, bio-degradation, photocatalysis, adsorption and chemical oxidation, have been used for the removal of dye effluents from waste water.3–6 Among these methods, adsorption is a simple and effective technology. Many kinds of materials such as activated carbon, clay minerals, alumina and molecular sieves, etc. have been used as adsorbents to remove dye pollutants from waste water.7–11 However, the wide application of these aforementioned adsorbents is limited by their relatively low adsorption capacities, difficult regeneration and/or high-cost. Therefore, it is still of great interest to fabricate environmentally compatible and efficient functional adsorbents for dye removal.
Layered double hydroxides (LDHs), known as hydrotalcite-like compounds, can be expressed as [M1−x2+Mx3+(OH)2](An−)x/n·mH2O, where M2+ is a divalent cation like Mg2+, Zn2+, Cu2+, etc., M3+ is a trivalent cation like Al3+, Fe3+, etc., and An− is the intercalated anion with n− charge. By virtue of their versatility in morphology, composition and architecture, LDHs have been studied as catalysts, catalyst promoters, especially adsorbents and ion-exchangers for removal of many anionic organic pollutants in water.12–14 Particularly worth mentioning is that the calcination of magnesium aluminum LDH can lead to the formation of periclase-like Mg, Al-oxide solid solution. The obtained calcined LDH can recover the lamellar structure upon treatment with water or water solution containing anions.15,16 That is, when the calcined LDH is put into an aqueous solution for a certain time, the layered structure is reconstructed, admitting OH− or other anions in the solution as charge-compensating anions. This property is known as “memory effect”, which has been exploited for various applications of LDHs such as adsorption, anion exchange, catalysis, etc.16,17
LDHs are traditionally prepared by co-precipitation of metallic salts with an alkaline solution.18,19 Recently, some alternative routes have been explored in order to achieve desirable microstructure and texture of LDHs. For instance, Valente et al. employed a sol–gel method using ethanol, 2-propanol, and 1-butanol as solvents to synthesis LDHs with nanocapsular morphology, and calcined LDHs with large specific surface areas (254–332 m2 g−1).15,20 Chang et al. reported a ultrasound-assisted co-precipitation method in combination with a calcination treatment to prepare magnetic Mg–Al layered double hydroxides composite, and used it as an adsorbent to remove fluoride ions from water.21 Benito et al. used a microwave-hydrothermal method to obtain small size and monodispersed hexagonal particles of LDH compounds with specific surface area of 117 m2 g−2.22 Wang et al. also successfully synthesized ethylene glycol intercalated magnesium–aluminum layered double hydroxide via hydrothermal and ion exchange method.23 Different preparation routes can lead to difference of LDH in surface property, morphology and texture including pore size and specific surface area, and the subsequent adsorption performance for dye removal. LDH prepared by a microemulsion method, to the best of our knowledge, has not yet been studied. The attractiveness of microemulsion-based method is capable to control the properties of the synthesized nanomaterials such as particle size, surface area, morphology and homogeneity.24–26
Orange II (O-II) is known as one of toxic and carcinogenic azo dyes. Herein, O-II is chosen as a model pollutant since it is inexpensive and widely used in the textile, leather and paper industries. The chemical structure of O-II is shown as Fig. 1. In the present work, we used a microemulsion method at a relatively low temperature (70 °C) to prepare magnesium–aluminum layered double hydroxide and oxide with large specific surface area, and used the obtained adsorbents to remove O-II dye from water.
 |
| | Fig. 1 The chemical structure of orange II. | |
Experimental
Preparation of adsorbents
Mesoporous LDH was synthesized by a water-in-oil microemulsion method; the preparation process was reported in our previous study in detail.24–26 Briefly, a mixture containing 100 mL cyclohexane and 20.4 g polyethyleneglycol (PEG 400) was magnetically stirring and heated to 70 °C. Then 20 mL solution containing Mg(NO3)2·6H2O (0.005 M) and Al(NO3)3·9H2O (0.005 M) was added to the mixture. After stirring about 10 min, 3.5 g NH3 solution (27%) was added to the above solution. The microemulsion was then aged for 2 h to generate precipitation. The obtained precipitation was separated by centrifuging, and washed five times with water and ethanol, respectively, and then dried overnight at ca. 80 °C.
Preparation of calcined LDH: the as-prepared LDH was annealed in air at 300, 400, 500, 600 °C for 2 h at a heating rate of 2 °C min−1, and denoted as LDH-300, LDH-400, LDH-500 and LDH-600, respectively.
Characterization
X-ray diffraction (XRD) patterns of the samples were obtained using a Philips X'Pert powder X-ray diffractometer with Cu Kα radiation (λ = 0.15419 nm). Nitrogen adsorption–desorption isotherms were obtained on an ASAP 2020 (Micromeritics Instruments, USA). All samples were degassed at 150 °C prior to adsorption measurements. The Brunauer–Emmett–Teller (BET) surface area (SBET) was determined by a multipoint BET method by using adsorption data in the relative pressure P/P0 range of 0.05–0.2. The single-point pore volume (Vp) was obtained from the amount adsorbed at a relative pressure of 0.98. The pore size distribution (PSD) was calculated using adsorption branches of nitrogen adsorption–desorption isotherms. Transmission electron microscopy (TEM) measurement was performed on a JEM-2100F electron microscope (JEOL, Japan). Thermogravimetric analysis (DTA-TGA) was performed on a DTG-60H analyzer (Shimadzu Corp., Tokyo, Japan) in air using a heating rate of 5 °C min−1. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Kratos XSAM800 XPS system with Al Kα source and a charge neutralizer, and all the binding energies were referenced to the C 1s peak at 284.8 eV of the surface adventitious carbon. Fourier transform infrared spectra (FTIR) were collected using a Shimadzu IRAffinity-1 FTIR spectrometer in a range of 400–4000 cm−1. The zeta potentials of the samples were measured at 25 °C using a zeta potential analyzer (Malvern, Zetasizer Nano, ZS90).
Adsorption experiment
The adsorption isotherm measurements were performed by adding 10 mg of the as-prepared adsorbents to a series of beakers containing 200 mL of diluted orange-II (C16H11N2NaO4S, O-II) solutions (5–40 mg L−1). The beakers were sealed and kept at air-bath oscillator under 30 °C for 72 h to reach equilibrium. The equilibrium concentration of O-II was measured at the maximum absorption wavelength of O-II (484 nm) using a UV-vis spectrophotometer (UV-1240, Shimadzu, Japan). The amount of O-II adsorbed at equilibrium (qe, mg g−1) was calculated using the following equation:| |
 | (1) |
where C0 and Ce (mg L−1) are the initial and equilibrium concentrations of O-II, respectively, V is the volume of the solution (L), and W is the mass of adsorbent used (g).
The adsorption kinetic tests were similar to the adsorption isotherm measurements. Adsorption kinetics of O-II was investigated at 30 °C in an air-bath oscillator. In a typical experiment, the initial concentration of the pollutant is 30 mg L−1 and the dosage of the adsorbent is 50 mg L−1. Analytical samples were taken from the suspension after at pre-set time intervals and separated by centrifuging. The amount of the pollutant dye adsorbed at time t (qt, mg g−1) was calculated by:
| |
 | (2) |
where
C0 and
Ct (mg L
−1) are the concentrations of O-II at initial and specified time
t, respectively,
V is the volume of the solution (L), and
W is the mass of adsorbent used (g).
Results and discussion
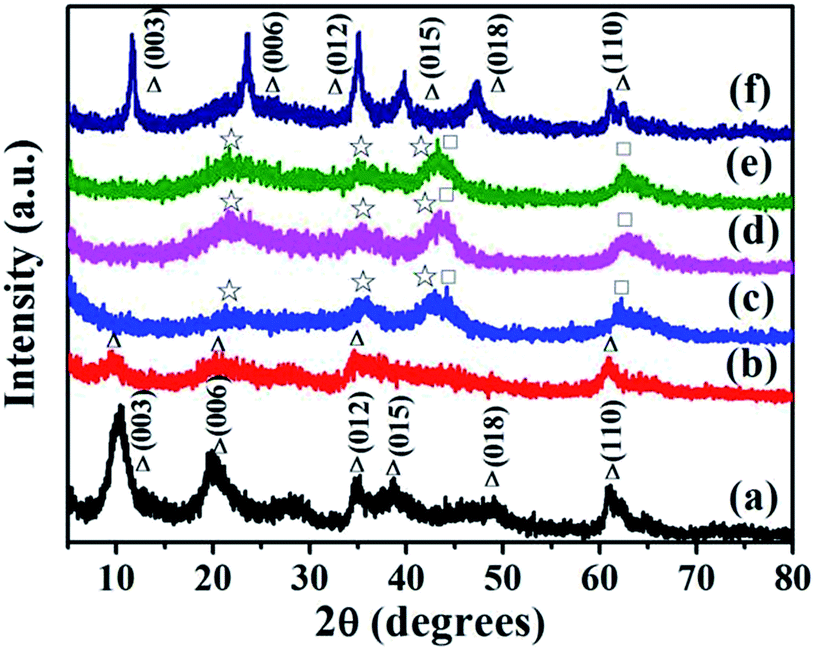
Typical XRD patterns of the studied samples are shown in Fig. 2. The peaks appear at 2θ = 10.4, 20.2, 34.7, 38.5, 49.1 and 61° in the XRD pattern of the as-prepared LDH, corresponding to (003), (006), (012), (015), (018) and (110) lattice planes, respectively. It indicates the formation of a typical lamellae structure.27–31 The interlayer space region may contain some anions such as OH−, NO3− and CO32− to balance the excess of positive charge within the Mg(Al)–O layers. Compared to those reported in the literatures,30,31 the diffraction peaks of the LDH are much broader, implying that the obtained LDH consists of amorphous structure or its crystalline particles are smaller. In addition, the basal spacing calculated from the (003) peak of LDH is 8.51 Å. Some organic molecules like PEG or its derivative are reasonably inferred to intercalate into the layer of the obtained LDH since the sample is prepared in the microemulsion process, which can be confirmed by its TG and DTA result. LDH-300 shows less intensive peaks compared to the as-prepared LDH, mainly due to partial loss of interlayer water. For LDHs thermally calcined at above 400 °C, the peaks are distinct from those of LDH and LDH-300; the peaks indicative of the typical LDH lamellae structure disappear, suggesting an almost total decomposition of the original LDH. New phases manifested by the presence of new broad reflections appear for LDH-400, LDH-500 and LDH-600 samples. The positions of diffraction lines are indexed to the diffraction peaks of MgO (JCPDS no. 45-0946) and magnesium aluminum oxide hydrate (MgAl2O4·7.9H2O, JCPDS no. 22-0432). The result indicates that most of the intercalated ions can be removed at above 400 °C. After LDH-500 adsorbed O-II in the solution, a typical layered structure of the LDH was reconstructed (Fig. 2f); and the basal spacing (d003) value of the reconstructed LDH is ca. 7.63 Å, larger than that reported in the literature for intercalation of OH− ions or CO32− in Mg–Al-LDH.27,28 This implies that some O-II anions and OH−/CO32− ions simultaneously intercalate into the layers in the process of structural reconstruction. Moreover, compared to the as-prepared LDH (Fig. 2a), the 2θ of (003) and (006) peaks shift to higher values. This illustrates that the reconstruction structure of LDH-500 differs from that of the as-prepared LDH due to the different intercalated balance-ions.
 |
| | Fig. 2 XRD patterns of (a) LDH, (b) LDH-300, (c) LDH-400, (d) LDH-500, (e) LDH-600 and (f) LDH-500 after adsorption of O-II. (Δ hydrotalcite phase; ☆ magnesium aluminum oxide hydrate phase; □ MgO phase). | |
Generally, adsorbents with high specific surface area, large pore volume and appropriate pore sizes perform well due to high concentration of surface active sites that enhances adsorption of adsorbate molecules and facilitate their diffusion into the interior of adsorbents. Therefore, the pore structure and surface area of LDH and LDHs calcined under different temperature are investigated by N2 adsorption–desorption measurements. Fig. 3 shows the nitrogen adsorption–desorption isotherms and the corresponding pore-size distribution (PSD) curves of the as-prepared samples. All the isotherms can be classified as type IV, indicative of the presence of mesoporous structure. Moreover, all the samples show H2-type hysteresis loops, which are related to an interconnected pore network.32 The condensation step of the isotherm measured on LDH-500 and LDH-600 shifts toward higher P/P0 as compared to those of LDH, LDH-300 and LDH-400. This suggests that LDH-500 and LDH-600 have larger mesopores, which can be confirmed by the PSD curves. The PSD curves for LDH-500 and LDH-600 show a peak located at the pore diameter of 12.1 nm and 14.3 nm, respectively; while the peaks for LDH, LDH-300 and LDH-400 are centered at 3.0, 2.5 and 4.1 nm, respectively. The result shows that the peak mesopore increases with increasing calcination temperature. The SBET, Vp, and pore size width (dp) at the maximum of PSD for the as-prepared samples are listed in Table 1. LDHs thermally treated at higher temperature exhibit larger specific surface areas and pore volumes, mainly due to the removal of interlayer water and intercalation ion of the starting material. Furthermore, the LDH-500 and LDH-600 show the highest specific surface area of ca. 315 m2 g−1, similar to the calcined LDH at 500 °C (332 m2 g−1) prepared by sol–gel method using 1-butanol as solvent,20 and much larger than the reported calcined LDH (219 m2 g−1).33 This indicates an advantage of the microemulsion synthesis. A larger surface area can offer more active adsorption sites, which might potentially induce a higher organic pollutant adsorption capacity.
 |
| | Fig. 3 The nitrogen adsorption–desorption isotherms (A) and the corresponding PSD curves (B) of the as-prepared samples. | |
Table 1 Textural properties of the samples studied
| Samples |
SBET (m2 g−1) |
dp (nm) |
Vp (cm3 g−1) |
| LDH |
161 |
3.0 |
0.26 |
| LDH-300 |
212 |
2.5 |
0.30 |
| LDH-400 |
231 |
4.1 |
0.43 |
| LDH-500 |
315 |
12.1 |
0.96 |
| LDH-600 |
316 |
14.3 |
1.06 |
The TEM images are recorded to observe the morphologies of LDH and LDH-500 (shown in Fig. 4). The TEM image of LDH presents a porous microstructure composed of randomly aggregated and interconnected ultrathin nanoflakes. Compared to those prepared via conventional co-precipitation or sol–gel method,34 the nanoflakes of the obtained LDH here are much thinner and smaller, indicative of a restricting effect of microemulsion. LDH-500 still exhibits the morphology of aggregated nanoflakes after calcined at 500 °C, implying that calcination did not change the morphology. Closer observation is found that the aggregated nanoflakes of LDH-500 sample are smaller and discrete compared to those of LDH sample, which is caused by the loss of structure water and intercalated ions for LDH-500 sample. The loss of structure water and intercalated ions would result in more porous microstructure of LDH-500 and/or larger specific surface area, which can be verified by the result of the nitrogen adsorption–desorption isotherms.
 |
| | Fig. 4 TEM images of LDH (a) and LDH-500 (b). | |
Fig. 5 shows TG and DTA curves of LDH and LDH-500. The TG curves of the two samples are divided into three mass loss steps. For LDH, the loss in the first region from 50 to 150 °C is ca. 8%, resulting from the desorption of physisorbed water.35 In the second region between 150 and 500 °C, the weight loss is ca. 35%, mainly attributed to the removal of the intercalated ions and organic molecule like PEG. A big exothermic peak at ca. 260 °C in DTA curve is related to the combustion of polyethyleneglycol trapped in the nanostructures,36 and a small exothermic peak at ca. 400 °C is ascribed to the combustion of polyethyleneglycol intercalated in the layered structure of LDH. The final loss from 500 to 700 °C is ca. 3%, corresponding to continuous elimination of strongly bounded anions. LDH-500 exhibits different DTA and TG curves from those of LDH. In the first step, the weight loss is ca. 9% due to the loss of surface adsorbed water. The weight loss in the second step is ca. 11%, due to the loss of chemisorbed water and/or partial structure water, and the loss in the final step is ca. 3% related to continual loss of remnant intercalated anions. Compared to LDH, LDH-500 shows a less weight loss in the second step; no obvious exothermic peaks related to the combustion of organic pollutant are observed. This indicates that the organic substance and most of the intercalated ions are removed when the LDH was calcinated at 500 °C.
 |
| | Fig. 5 TG and DTA profiles of LDH and LDH-500. | |
XPS was used to analyze the elemental composition of the samples. Fig. 6a shows survey XPS spectra of LDH and LDH-500 samples. Spectrum peaks associated with Mg, Al, O and C are observed for the two samples, indicative of the successful preparation of magnesium aluminum LDH. N element is observed only on the spectrum of LDH. It suggests the existence of LDH-NO3. Moreover, the result also implies that the intercalated NO3− ions in LDH can be removed at 500 °C.
 |
| | Fig. 6 XPS spectra of the survey (a), O 1s (b) and C 1s (c) in LDH and LDH-500. | |
The XPS spectra of O 1s (shown in Fig. 6b) are divided into three peaks at 531.1–531.2, 532–532.1 and 533.2 eV, which are attributed to the oxygen in lattice O, OH−/CO32− and adsorbed water, respectively.34 The ratio of oxygen in OH−/CO32− for LDH is ca. 62.4%, much larger than that for LDH-500 (39%). Moreover, the oxygen in adsorbed water for LDH is ca. 6.6%, smaller than that for LDH-500 (10.7%), indicating the strong affinity of LDH-500. The asymmetric C 1s spectra of the two samples (shown in Fig. 6c) can be deconvoluted into three peaks at 284.7–284.8, 286.2–286.4, and 288.7–289 eV, which are attributed to the adventitious hydrocarbon from the XPS instrument itself, the C in C–O bonds and the carboxylate C (CO32−), respectively.37 Combining the analysis result of the O 1s with C 1s, it can be concluded that Mg2+ is linked by both oxide/hydroxide and CO32−.
Fig. 7 shows the FTIR spectra of LDH and LDH-500 before and after O-II adsorption. A strong broad band centered at ca. 3470 cm−1 and a small peak at ca. 1630 cm−1 in the spectra of all the samples are associated with the stretching and bending vibrations of the hydrogen-bonded hydroxyl groups from the surface and/or interlayer, respectively. The bands at 1068 and 1390 cm−1 are assigned to monodentate carbonate.38 In addition, the stronger peak at ca. 1390 cm−1 is also related to the stretching vibration of NO3− intercalated in the interlayer space for LDH sample. Compared to LDH and LDH-500, some new peaks appear in the FTIR spectra of LDH and LDH-500 after O-II adsorption. The band at 1512 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration, and the peak at 1186 cm−1 is due to the symmetric vibration of –O–S–(O2) group.39 Moreover, the peaks at 1122 and 1033 cm−1 are related to the coupling between the benzene mode and νs(SO3) (Fig. 7, inset).40 This result suggests O-II adsorption onto the LDH and LDH-500 samples.
C stretching vibration, and the peak at 1186 cm−1 is due to the symmetric vibration of –O–S–(O2) group.39 Moreover, the peaks at 1122 and 1033 cm−1 are related to the coupling between the benzene mode and νs(SO3) (Fig. 7, inset).40 This result suggests O-II adsorption onto the LDH and LDH-500 samples.
 |
| | Fig. 7 FTIR spectra of (a) LDH, (b) LDH-500, (c) LDH after O-II adsorption and (d) LDH-500 after O-II adsorption. Inset shows the amplified spectrum between 1000 and 1200 cm−1. | |
To compare the surface charges of LDH and LDH-500 samples, their zeta potentials were measured as shown in Fig. 8. The zeta potentials continuously decrease as the pH increases, and the isoelectric points (IEPs) of LDH and LDH-500 are about 11.0 and 10.4, respectively. The LDH and LDH-500 are positively charged at pH values below the IEP and negatively charged above this point. It suggests that the two samples are positively charged in the studied solution (pH = 7). Therefore, at the pH studied, O-II is expected to be adsorbed on the surface of the as-prepared samples due to electrostatic attraction between the positively charged adsorbents and sulfonic acid groups of the O-II molecules.
 |
| | Fig. 8 Zeta potentials of LDH and LDH-500 as functions of the pH value of the suspensions. | |
The investigation of adsorption kinetics is significant for the removal processes of organic pollutant since it can evaluate the adsorption rate at which the pollutant is removed from aqueous solutions and provide valuable data for understanding the adsorption mechanism.3,41 The effect of contact time on O-II removal by LDH, LDH-300, LDH-400, LDH-500 and LDH-600 is shown in Fig. 9. LDH, LDH-300 and LDH-400 exhibited a quite fast adsorption rate at the initial 250 min, while LDH-500 and LDH-600 showed a quite fast adsorption rate at the first 400 min. Afterward, the adsorption rates on all the samples gradually became slower against contact time with the final quasi-equilibrium being achieved within ca. 450 minutes for LDH, LDH-300 and LDH-400; and ca. 600 minutes for LDH-500 and LDH-600. It can be observed that the LDH-500 and LDH-600 samples had the largest adsorption capacity, which was two times higher than that of the as-prepared LDH. The much larger O-II uptake is attributed to the larger specific surface areas and pore volumes, and strong adsorption affinity of LDH-500 and LDH-600 resulted from their peculiarly structural reconstruction. O-II molecules were presumed to diffuse into the interior and interlayer structure of LDH-500 and LDH-600 reconstruction, which was accompanied by a longer equilibrium time and a higher O-II uptake.
 |
| | Fig. 9 Effect of contact time on adsorption of O-II on LDH, LDH-300, LDH-400, LDH-500 and LDH-600 samples (T = 30 °C, adsorbent dose = 50 mg L−1, O-II concentration = 30 mg L−1 and pH = 7). | |
The adsorption kinetics was further studied using the pseudo-first-order and pseudo-second-order kinetic models, and the corresponding linear forms of the kinetic equations are as follows:
| |
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (3) |
| |
 | (4) |
where
qe and
qt are the amounts of O-II adsorbed (mg g
−1) per unit of adsorbent at equilibrium and specified time
t (min), respectively;
k1 and
k2 are the pseudo-first-order and pseudo-second-order rate constants, respectively. For quantitative evaluation of the suitable kinetic model, correlation coefficients (
R2) were compared. The kinetic studies revealed that the pseudo-second-order equation was well fitted with the experimental data due to the high values of
R2 (shown in
Fig. 10); the calculated kinetic parameters were listed in
Table 2. This result suggests that the rate control mechanism of adsorption is chemical adsorption.
42 Similar phenomena are also observed for other types of dyes and contaminants adsorbed on calcined LDHs.
43–45
 |
| | Fig. 10 Pseudo-second-order kinetics for adsorption of O-II on LDH, LDH-300, LDH-400, LDH-500 and LDH-600 samples (T = 30 °C, adsorbent dose = 50 mg L−1, O-II concentration = 30 mg L−1 and pH = 7). | |
Table 2 Pseudo-second-order adsorption kinetic constants of the samplesa
| Samples |
C0 (mg L−1) |
qe,exp (mg g−1) |
Pseudo-second-order model |
| qe,cal (mg g−1) |
k2 (×10−6 g mg−1 min−1) |
R2 |
| qe,exp was obtained at the adsorption time of 36 h. |
| LDH |
30 |
136 |
140 |
110.79 |
0.997 |
| LDH-300 |
30 |
74 |
74 |
201.13 |
0.995 |
| LDH-400 |
30 |
131 |
111 |
74.52 |
0.995 |
| LDH-500 |
30 |
395 |
402 |
11.52 |
0.993 |
| LDH-600 |
30 |
382 |
535 |
3.83 |
0.960 |
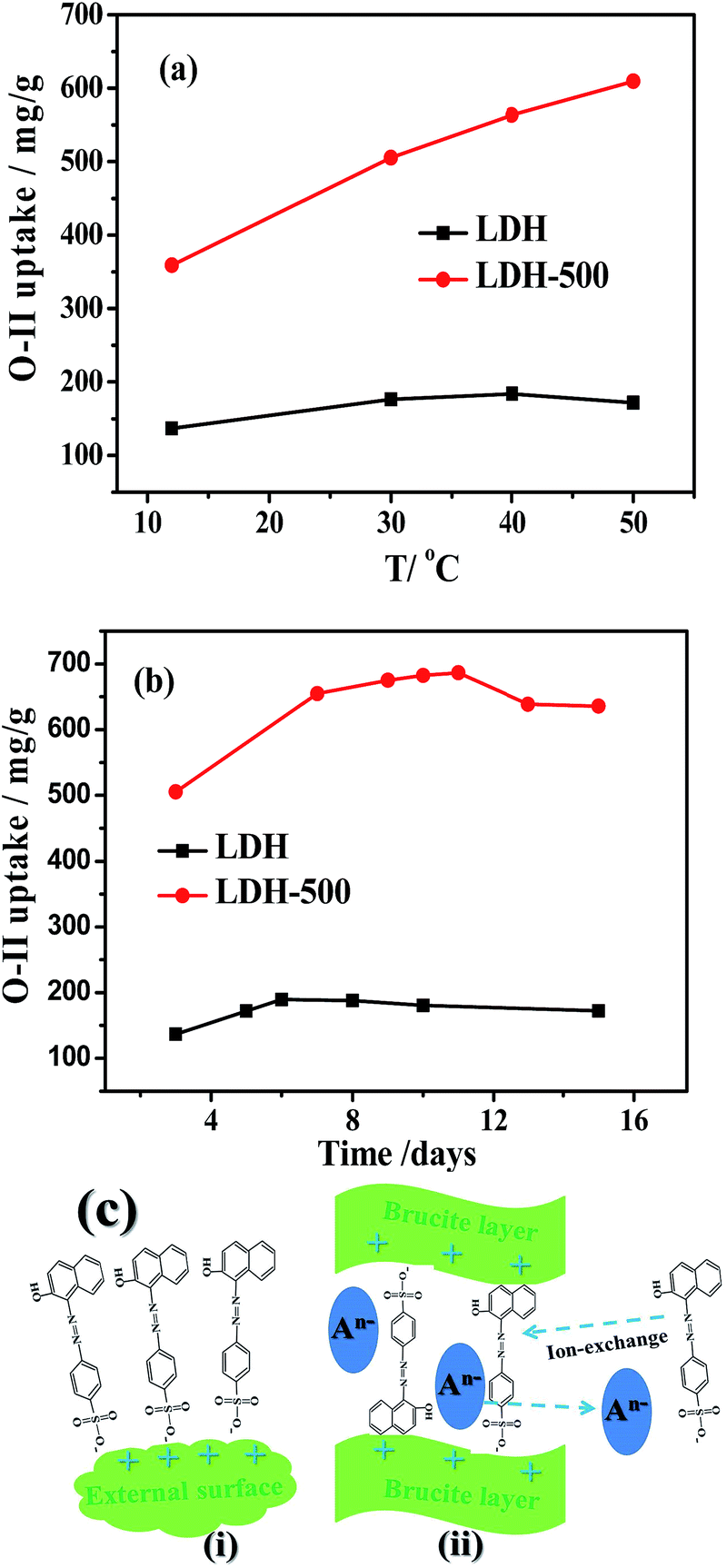
Fig. 11a shows the relationship between O-II uptake and temperature on LDH and LDH-500. For LDH, the O-II uptake increased with increasing temperature and reached the maximum at 40 °C, after which the adsorption capacity decreased. For LDH-500, the trend was different. The O-II uptake continuously increased with the increase of the temperature, demonstrating that a higher temperature was beneficial for increasing the O-II adsorption on LDH-500. It is easy to understand that the rising temperature speeded up the diffusion of O-II molecules from the bulk solution into the interior and interlayer of the adsorbent and enhanced their accessibility to the adsorbent active sites.46
 |
| | Fig. 11 Effect of temperature (a) and long contact time (b) on O-II uptake on LDH and LDH-500 adsorbents. Adsorbent dosage: 50 mg L−1; initial O-II concentration: 40 mg L−1. For (a) the equilibrium time is 3 days; for (b) the adsorption temperature is 30 °C. (c) Possible mechanism of O-II adsorption on the as-prepared samples. (i) Direct adsorption on external surface via electrostatic attraction and (ii) intercalation via ion-exchange (for LDH-500, direct intercalation in the reconstruction is not shown here). | |
The effect of long contact time on O-II removal by LDH and LDH-500 is shown in Fig. 11b. For LDH, the amounts of O-II adsorbed continuously increased within the first 6 days, and then slightly decreased after the 8th day. For LDH-500, the amounts of O-II adsorbed increased rapidly within initial 7 days and followed by a slow increase in the following 4 days, and then decreased at the end of the test. The increase of O-II uptake with increasing contact time at first (shown in Fig. 11b) is mainly due to the slow diffusion of O-II into the interlayer of the adsorbents via ion-exchange. Compared to LDH, LDH-500 showed much larger O-II uptake and longer adsorption equilibrium time. It is reasonable to assume that the slow diffusion of O-II into the interior of the adsorbent and the interlayer of LDH-500 reconstruction resulted in the larger O-II uptake and longer equilibrium time, which is similar to the results reported by Zhu et al. that Brilliant Blue R (BBR) dye was adsorbed on the calcined layered double hydroxides (CLDHs) via the BBR molecules intercalation into the reconstructed CLDHs.27
To evaluate the importance of the surface charge on adsorption of the dye pollutants, the adsorption experiments of methylene blue (MB) were performed using the as-prepared samples as adsorbents. The MB concentrations demonstrates negligible changes on all the adsorbents during the whole studied time of 750 min (not shown here), which is distinctly different from O-II concentration decreasing quickly within a short contact time. Based on the above results, we presume that the interaction mechanism in the adsorption of O-II onto the as-prepared samples may mainly involve (i) direct adsorption on external surface via electrostatic attraction and (ii) intercalation via ion-exchange (shown in Fig. 11c). Moreover, the direct intercalation of O-II into the reconstruction of LDH during the rehydrated process of LDH-500 plays an important role in its larger adsorption capacity of O-II.
Adsorption isotherms for O-II uptake on LDH and LDH-500 at 30 °C are shown in Fig. 12. Compared to that before adsorption, the color of O-II solutions with low concentrations after adsorption evidently faded (not shown), indicating the efficient removal of O-II from aqueous solution by LDH and LDH-500. Closer observation is found that LDH-500 exhibited a better adsorption ability than LDH. For example, at a similar O-II equilibrium concentration of 14 mg L−1, the adsorption capacity of LDH-500 was more than 4 times larger than that of LDH. It can be observed from Fig. 12 that LDH-500 can be used to remove anionic dyes of relatively high concentrations, while LDH may only be used to remove anionic dyes of low concentrations. Since the dye concentration usually ranges from 10 to 50 mg L−1 in most wastewater,27 LDH-500 could be a highly effective adsorbent. The larger adsorption capacity of LDH-500 is attributed to its peculiar property of structural reconstruction, higher surface area, larger pore size and strong affinity.
 |
| | Fig. 12 Adsorption isotherms for O-II on LDH and LDH-500 at 30 °C. Dosage of sorbents: 50 mg L−1, initial O-II concentration: 10, 15, 20, 25, 30, 35 and 40 mg L−1; equilibration time: 3 days. | |
The equilibrium adsorption data was analyzed using Langmuir isotherm equation as follows.9
| |
 | (5) |
where
Ce is the equilibrium concentration of O-II in solution,
qe is the amount of O-II adsorbed on the adsorbent at equilibrium (mg g
−1),
qmax is the maximum adsorption capacity of the adsorbent corresponding to the complete monolayer coverage (mg g
−1), and
KL is the Langmuir adsorption constant, which is related to the Gibbs free energy of adsorption. It is worth noting that the original Langmuir equation has been obtained for monolayer localized adsorption from an ideal gas phase on a homogeneous surface. The lateral interactions are neglected and adsorption is assumed on the unoccupied adsorption sites in this model. Although solute adsorption from dilute aqueous solutions might be expressed by the Langmuir equation, the adsorption mechanism is different from that at the gas/solid interface. For solute adsorption, there is adsorption competition between solute and solvent molecules, and the obtained
KL constant depends on the difference of solute and solvent adsorption energies.
9 Fig. 13 shows that the Langmuir adsorption isotherm is consistent with the experimental data, and its linear form can be derived from the
eqn (5). The fitted parameters are listed in
Table 3. The result showed that the adsorption of large dye molecules such as O-II was represented well by monolayer localized adsorption model. It is noteworthy that the maximum adsorption capacity of O-II dye on LDH-500 and LDH is 602 mg g
−1 and 224 mg g
−1, respectively, much larger than that on carbon–alumina spheres (209 mg g
−1)
9 and on modified palygorskites (88–92 mg g
−1).
11 This is attributed to the higher surface area, larger pore size and unique structure of LDH and LDH-500.
 |
| | Fig. 13 Langmuir linear plots of adsorption isotherms for O-II onto the LDH and LDH-500 at 30 °C. | |
Table 3 Adsorption isotherm parameters of the samples studied
| Sample |
qmax (mg g−1) |
KL (L mg−1) |
R2 |
| LDH |
224 |
0.144 |
0.989 |
| LDH-500 |
602 |
0.363 |
0.931 |
Thermodynamic parameters like the Gibbs free energy (ΔGo), enthalpy (ΔHo) and entropy (ΔSo) can be evaluated by the following equation.47
| |
 | (6) |
where
KL is the Langmuir adsorption constant,
R and
T is the gas constant (8.314 J mol
−1 K
−1) and temperature of the reaction, respectively. Δ
Go, Δ
Ho and Δ
So were derived from the slope and intercept of van't Hoff plot of ln
KL versus 1/
T (as shown in
Fig. 14 and
Table 4).
48 The positive value of Δ
Ho (28.7 kJ mol
−1) confirms the endothermic nature of the adsorption of O-II onto LDH-500, indicating that the higher temperature facilitates O-II adsorption on LDH-500. Thus, it is easy to understand the adsorption capacity of LDH-500 for O-II continuously increased with increasing temperature. The positive Δ
So (100 J mol
−1 K
−1) suggests the increased in randomness at the solid–solute interface during the adsorption process.
49 However, the low value of Δ
So also implies that no remarkable changes in entropy occurred during the adsorption. The value of Δ
Go decreased from 0.4 to −3.4 kJ mol
−1 with the temperature increasing from 12 to 50 °C, further illustrating the adsorption of O-II on LDH-500 was feasible and thermodynamically spontaneous at higher temperature.
 |
| | Fig. 14 Plot of lnKL vs. 1/T for O-II adsorption on LDH-500. Initial O-II concentration: 40 mg L−1. | |
Table 4 Thermodynamic parameters for O-II adsorption over LDH-500. Equilibrium time: 3 days
| |
ΔHo (kJ mol−1) |
ΔSo (J mol−1 K−1) |
ΔGo (kJ mol−1) |
| 12 °C |
30 °C |
40 °C |
50 °C |
| LDH-500 |
28.7 |
100 |
0.4 |
−1.5 |
−2.5 |
−3.4 |
Re-use is practically important for an adsorbent to remove pollutants from water. A recycling experiment for LDH-500 to adsorb O-II in water was performed (shown in Fig. 15). Calcined LDHs are usually capable of being regenerated due to the “memory effect”.27 By thermally treated at 500 °C, organic dye adsorbed on calcined LDHs can be almost completely decomposed, and the used calcined LDHs is regenerated again. As shown in Fig. 15, the adsorption capacity of LDH-500 for O-II decreased during the first two runs; the similar phenomenon was also observed on BBR adsorbed on calcined LDHs reported by Zhu et al.27 Then the O-II uptake on LDH-500 became relatively stable in the subsequent runs, indicating LDH-500 could be efficiently reused. The reduced adsorption capacity at the first two cycles probably resulted from the progressively decreasing crystallinity of the LDH-500 in structural reconstruction after thermal regeneration.12 After the first generation, the color of the thermally treated LDH-500 turned pale compared to pure white color of the as-prepared LDH-500, mainly due to a part of decomposed intermediates of O-II were incorporated into the regenerated LDH-500. Incorporation of O-II intermediates presumably disturbed the structural reconstruction of the LDH-500 and subsequently reduced its crystallinity and adsorption capacity.
 |
| | Fig. 15 O-II adsorption on LDH-500 as a function of recycle time. Adsorbent dosage: 50 mg L−1; initial O-II concentration: 40 mg L−1; equilibrium time: 3 days. | |
Conclusions
Calcined LDHs nanoflakes with large specific surface area were prepared by microemulsion and calcination processes. Calcined LDH at 500 °C presented the maximum adsorption capacity (602 mg g−1), which is much higher than that of LDH (224 mg g−1), mainly due to its larger specific surface area, larger pore size and pore volume as well as higher adsorption affinity resulted from its structural reconstruction. The pseudo-second-order kinetic and Langmuir isothermal models are best consistent with the experimental data. The adsorption temperature and long contact time have an obvious influence on O-II uptake for LDH and LDH-500. The adsorption of O-II dye by the as-prepared samples was mainly via an electrostatic interaction between anionic O-II molecules and positively charged surface of the adsorbents, and the slow intercalation of O-II into the layers of LDH and LDH-500 restructure. Owing to its unique structural reconstruction, high surface area and environment-friendliness, calcined LDH is potentially applicable in wastewater treatment.
Acknowledgements
This work was partially supported by the 973 Program (2013CB632402), NSFC (21307038, 21577046, 21433007, 51320105001 and 51272199), Outstanding Young Science and Technology Innovation Team Program of Hubei Provincial Universities (T201514), the Fundamental Research Funds for the Central Universities (2015-III-034), Self-determined and Innovative Research Funds of SKLWUT (2015-ZD-1) and the Natural Science Foundation of Hubei Province of China (No. 2015CFA001).
Notes and references
- R. Caritá and M. A. Marin-Morales, Chemosphere, 2008, 72, 722 CrossRef PubMed.
- R. O. A. Lima, A. P. Bazo, D. M. F. Salvadori, C. M. Rech, D. P. Oliveira and G. A. Umbuzeiro, Mutat. Res., 2007, 626, 53 Search PubMed.
-
(a) L. Gan, S. Shang, E. Hu, C. W. Yuen and S. Jiang, Appl. Surf. Sci., 2015, 357, 866 CrossRef CAS;
(b) R. Chen, J. Yu and W. Xiao, J. Mater. Chem. A, 2013, 1, 11682 RSC;
(c) C. Lei, X. Zhu, B. Zhu, J. Yu and W. Ho, J. Colloid Interface Sci., 2016, 466, 238 CrossRef CAS PubMed;
(d) J. Yu, X. Li, Z. Xu and W. Xiao, Environ. Sci. Technol., 2013, 47, 9928 CrossRef CAS PubMed;
(e) C. Lei, X. Zhu, Y. Le, B. Zhu, J. Yu and W. Ho, RSC Adv., 2016, 6, 10272 RSC;
(f) X. Yin, F. Zhang and W. Zhang, Appl. Surf. Sci., 2015, 359, 714 CrossRef CAS.
- Q. Q. Liu, L. Wang, A. G. Xiao, J. M. Gao, W. B. Ding, H. J. Yu, J. Huo and M. Ericson, J. Hazard. Mater., 2010, 181, 586 CrossRef CAS PubMed.
-
(a) R. Wang, X. Cai and F. Shen, Appl. Surf. Sci., 2011, 305, 352 CrossRef;
(b) D. Xu, B. Cheng, J. Zhang, W. Wang, J. Yu and W. Ho, J. Mater. Chem. A, 2015, 3, 20153 RSC;
(c) L. Cai, X. Xiong, N. Liang and Q. Long, Appl. Surf. Sci., 2015, 353, 939 CrossRef CAS;
(d) S. Song, B. Cheng, N. Wu, A. Meng, S. Cao and J. Yu, Appl. Catal., B, 2016, 181, 71 CrossRef CAS;
(e) Y. Duan, Q. Wen, Y. Chen, T. Duan and Y. Zhou, Appl. Surf. Sci., 2015, 320, 746 CrossRef.
- A. K. Verma, R. R. Dash and P. Bhunia, J. Environ. Manage., 2012, 93, 154 CrossRef CAS PubMed.
- P. C. C. Faria, J. J. Mórfão and M. F. R. Pereira, Water Res., 2004, 38, 2043 CrossRef CAS PubMed.
-
(a) O. Gok, A. S. Ozcan and A. Ozcan, Appl. Surf. Sci., 2010, 256, 5439 CrossRef;
(b) Y. Le, D. Guo, B. Cheng and J. Yu, Appl. Surf. Sci., 2013, 274, 110 CrossRef CAS.
-
(a) J. Zhou, C. Tang, B. Cheng, J. Yu and M. Jaroniec, ACS Appl. Mater. Interfaces, 2012, 4, 2174 CrossRef CAS PubMed;
(b) W. Yang, P. Ding, L. Zhou, J. Yu, X. Chen and F. Jiao, Appl. Surf. Sci., 2013, 282, 38 CrossRef CAS.
-
(a) M. Liu, J. Xu, B. Cheng, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 332, 121 CrossRef CAS;
(b) R. Y. L. Yeh and A. Thomas, J. Chem. Technol. Biotechnol., 1995, 63, 55 CrossRef CAS;
(c) B. Zhu, P. Xia, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 344, 188 CrossRef CAS.
- B. Sarkar, Y. Xi, M. Megharaj and R. Naidu, Appl. Clay Sci., 2011, 51, 370 CrossRef CAS.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124 CrossRef CAS PubMed.
- F. Silvério, M. J. Reis, J. Tronto and J. B. Valim, J. Mater. Sci., 2008, 43, 434 CrossRef.
- Y. Seida and Y. Nakano, Water Res., 2002, 36, 1306 CrossRef CAS PubMed.
- J. S. Valente, E. Lima, J. A. Toledo-Antonio, M. A. Cortes-Jacome, L. Lartundo-Rojas, R. Montiel and J. Prince, J. Phys. Chem. C, 2010, 114, 2089 CAS.
- L. P. Cardoso and J. B. Valim, J. Phys. Chem. Solids, 2004, 65, 481 CrossRef CAS.
- M. Cantu, E. Lopez-Salinas, J. S. Valente and R. Montiel, Environ. Sci. Technol., 2005, 39, 9715 CrossRef CAS PubMed.
- W. T. Reichle, Solid State Ionics, 1986, 22, 135 CrossRef CAS.
- T. Kameda, E. Kondo and T. Yoshioka, Sep. Purif. Technol., 2014, 122, 12 CrossRef CAS.
- J. S. Valente, M. S. Cantú, G. H. Cortez, R. Montiel, X. Bokhimi and E. López-Salinas, J. Phys. Chem. C, 2007, 111, 642 CAS.
- Q. Chang, L. Zhu, Z. Luo, M. Lei, S. Zhang and H. Tang, Ultrason. Sonochem., 2011, 18, 553 CrossRef CAS PubMed.
- P. Benito, F. M. Labajos and V. Rives, Cryst. Growth Des., 2006, 6, 1961 CAS.
- H. Wang, W. Duan, Y. Wu, Y. Tang and L. Li, Inorg. Chim. Acta, 2014, 418, 163 CrossRef CAS.
- Z. Xu, J. Yu, J. Low and M. Jaroniec, ACS Appl. Mater. Interfaces, 2014, 6, 2111 CAS.
- Z. Xu, J. Yu and W. Xiao, Chem.–Eur. J., 2013, 19, 9592 CrossRef CAS PubMed.
- Z. Xu, J. Yu and M. Jaroniec, Appl. Catal., B, 2015, 163, 306 CrossRef CAS.
- M. Zhu, Y. Li, M. Xie and H. Xin, J. Hazard. Mater., 2005, 120, 163 CrossRef CAS PubMed.
- R. M. M. Santos, R. G. L. Gonçalves, V. R. L. Constantino, L. M. Costa, L. H. M. Silva, J. Tronto and F. G. Pinto, Appl. Clay Sci., 2013, 80–81, 189 CrossRef.
- Y. Yang, G. Fan and F. Li, Mater. Lett., 2014, 116, 203 CrossRef CAS.
- H. Wang, W. Duan, Y. Wu, Y. Tang and L. Li, Inorg. Chim. Acta, 2014, 418, 163 CrossRef CAS.
- N. Touisni, F. Charmantray, V. Helaine, C. Forano, L. Hecquet and C. Mousty, Colloids Surf., B, 2013, 112, 452 CrossRef CAS PubMed.
- C. Sangwichien, G. L. Aranovich and M. D. Donohue, Colloids Surf., A, 2002, 206, 313 CrossRef CAS.
- P. Liu, M. Derchi and E. J. M. Hensen, Appl. Catal., A, 2013, 467, 124 CrossRef CAS.
- Y. Lin, Q. Fang and B. Chen, Chem. Eng. J., 2014, 237, 38 CrossRef CAS.
- J. S. Valente, J. Prince, A. M. Maubert, L. Lartundo-Rojas, P. Angel, G. Ferrat, J. G. Hernandez and E. Lopez-Salinas, J. Phys. Chem. C, 2009, 113, 5547 CAS.
- Z. Xu, J. Yu and G. Liu, Dalton Trans., 2013, 42, 10190 RSC.
- R. Nie, J. Wang, L. Wang, Y. Qin, P. Chen and Z. Hou, Carbon, 2012, 50, 586 CrossRef CAS.
- N. Chubar, V. Gerda, O. Megantari, M. Mičušík, M. Omastova, K. Heister, P. Man and J. Fraissard, Chem. Eng. J., 2013, 234, 284 CrossRef CAS.
- J. Bandara, J. A. Mielczarski and J. Kiwi, Langmuir, 1999, 15, 7670 CrossRef CAS.
- G. S. Zhang, J. H. Qu, H. J. Liu, A. T. Cooper and R. C. Wu, Chemosphere, 2007, 68, 1058 CrossRef CAS PubMed.
- R. C. Wu, J. H. Qu and Y. S. Chen, Water Res., 2005, 39, 630 CrossRef CAS PubMed.
- Y. Lin, H. Chen, P. Chien, C. Chiou and C. Liu, J. Hazard. Mater., 2011, 185, 1124 CrossRef CAS PubMed.
- L. A. Lv, W. Wang, M. Wei and H. J. Cheng, J. Hazard. Mater., 2008, 152, 1130 CrossRef CAS PubMed.
- H. Zaghouane-Boudiaf, M. Boutahala and L. Arab, Chem. Eng. J., 2012, 187, 142 CrossRef CAS.
- E. L. Crepaldi, J. Tronto, L. P. Cardoso and J. B. Valim, Colloids Surf., A, 2002, 211, 103 CrossRef CAS.
- M. Sun, Y. Xiao, L. Zhang, X. Gao, W. Yan, D. Wang and J. Su, Chem. Eng. J., 2015, 272, 17 CrossRef CAS.
- J. Fan, W. Cai and J. Yu, Chem.–Asian J., 2011, 6, 2481 CrossRef CAS PubMed.
- A. Özcan, E. M. Öncü and A. S. Özcan, Colloids Surf., A, 2006, 277, 90 CrossRef.
- X. Jin, B. Yu, Z. Chen, J. M. Arocena and R. W. Thring, J. Colloid Interface Sci., 2014, 435, 15 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.