
Copper-loaded hypercrosslinked polymer decorated with pendant amine groups: a green and retrievable catalytic system for quick [3 + 2] Huisgen cycloaddition in water†
Arash Mouradzadegun* and
Mahsa Alsadat Mostafavi
Department of Chemistry, Faculty of Science, Shahid Chamran University of Ahvaz, Iran. E-mail: arash_m@scu.ac.ir; Fax: +98 61 33337009; Tel: +98 61 33331042
First published on 25th April 2016
Abstract
A novel heterogeneous copper catalyst based on a 3D-network polymer containing pendant amine groups was synthesized via post-functionalization of the polymeric backbone followed by incorporation of copper(I) ions. Catalytic activity of the newly designed catalyst was tested in a one-pot synthesis of 1,4-disubstituted 1,2,3-triazoles by click reactions between primary halides, sodium azide and terminal acetylenes in water under aerobic conditions. It increases the reaction rate many fold when compared with common transition metal catalysts. The synergistic incorporation of the porous matrix and active sites of this new catalyst offered a unique combination for effective catalytic-substrate interactions, which made it a new class of solid-support transition metal catalysts for chemical preparation.
Introduction
Sustainable chemistry focuses on the design, manufacture, and use of chemicals and chemical processes that cause little or no pollution potential or environmental risk and are both economically and technologically feasible.1 Since optimization of existing chemical processes together with the development of novel, environmentally friendly processes depend greatly on improvement of catalyst performance; the catalysis has a strong impact on the development of modern sustainable chemistry.Recently, solid-support heterogeneous catalysis have drawn great interest in industrial processes owing to their potential advantages over solution-based homogeneous catalysis, such as the easier operation/purification and recycling catalysts from reaction environments which lead to improved processing steps, better process economics, and environmentally friendly manufacturing.2 However, compared with the similar homogeneous catalysis, the heterogeneous catalysis usually suffered from decreased reactivity owing to the less effective interactions between catalytic centers and reactants. This is challenging for transition metal-promoted organic transformations, in which a good mix between catalysts and substrates is often required for effective reactions.
In order to solve this problem, porous materials have been proposed as a new type of solid-support for heterogeneous catalysis with much higher efficiency in catalyst–reactant interactions.3 In this context, inorganic porous materials, such as mesoporous silica and zeolite have received considerable attention. However, defects such as need to expensive organic surfactants or templates and calcinations at high temperature for obtaining opening channels have restricted their widespread practical applications on an industry scale.
Compared with inorganic supports, porous organic polymers (POPs), a new class of porous materials with a cross-linked amorphous organic framework, have emerged as a sustainable catalyst support owing to their advantages in good dispersion of active sites, pore-surface modifiability and the adsorption of reactant molecules on the material surface.4 Moreover, owing to the inherited nature of the organic pores, the cavities of the organic polymers are the best mimic of the chemical environment in the organic solvent-mediated homogeneous catalysis and thus help in the maintenance of the catalyst reactivity. These features, coupled with their biodegradability and green nature, make POPs attractive materials to organic and industrial chemistry.
Although a variety of POPs have been reported, some of them suffer from one or more defects in their synthetic or functionalization routes and properties including long reaction times, multiple steps for synthesis, functionalization via copolymerization or further post-polymerization under harsh reaction conditions, the use of expensive catalysts, solubility in various solvents, and low thermal and/or chemical stability. Therefore, developing and applying new POPs for heterogeneous-type catalytic systems, which may quickly move from the laboratory to green industrial plants, have received increased attention.
3D-Network polymer based on calix[4]resorcinarene could be ideal candidate for the development of solid-support POPs due to both their simple synthesis and the unique properties such as straightforward functionalization route via post-functionalization without undergoing copolymerization or further post-polymerization, versatile chemical modification (acylation, alkylation and silylation of hydroxyl group and nucleophilic aromatic substitution on the aromatic rings) and high thermal and chemical stability.
Despite convenient properties and significant potential of 3D-network polymer based on calix[4]resorcinarene in functionalization under mild reaction conditions without the use of acidic or basic catalyst, so far, there have been no reports on the use of these porous organic polymer as heterogeneous organometallic catalyst.
1,2,3-Triazole derivatives, five membered nitrogen containing heterocycles, have received considerable interest because of their tremendous applications in various research fields including organic synthesis, material science and cell biology.5 The classical method for the synthesis of these derivatives, thermal 1,3-dipolar cycloaddition of azides with alkynes, suffers from a few disadvantages such as low yields and lack of selectivity. To overcome these drawbacks, the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) has rapidly crossed traditional disciplinary boundaries.6 Toxicity of copper ions to cells and organisms coupled with high cost of transition metal catalysts has led to an increasing interest in immobilizing these catalysts onto a support. This class of supported reagent can facilitate both the isolation and recycling of the catalyst by filtration, thus providing environmentally cleaner processes.
Due to extensive practical validation of this transformation, several examples of polymer-supported catalysts have been reported in the literature. However, in most of these cases, nonporous polymer and porous polymers with low porosity and surface area were used generally which limited the interaction between substrates and catalytic sites. Furthermore, most of the reliable CuAAC protocols are performed under anaerobic conditions. These defects coupled with the need to extended reaction times and the use of organic solvents render the approaches unsatisfactory from the standpoint of green chemistry.
Under this light, as part of our continuing efforts to develop high performance and environmentally friendly procedures for various important reactions and transformations7 and due to our new interest in the application of the 3D-network porous polymer based on calix[4]resorcinarene for the preparation of biologically important molecules,8 herein, we have attempted to design, for the first time, a new type of porous network polymer containing a high concentration of amine sites as a more effective heterogeneous catalyst in promoting CuAAC reaction under aerobic conditions in water.
Experimental
Chemicals were purchased from Fluka, Merck and Aldrich chemical companies and used without further purification. Products were characterized by physical data, IR, 1H NMR, 13C NMR and HRMS spectra. Melting points were determined on a Thermo Scientific 9200 apparatus. IR spectra were obtained on a Bomen MB:102 FT-IR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on a Bruker spectrometer at 400 and 100 MHz, respectively, in CDCl3 or DMSO with tetramethylsilane as an internal standard. HRMS spectra were measured on an Agilent 5975 mass spectrometer and elemental analyses were performed on a Thermo Finnigan Flash EA 1112 CHNS-Analyzer. The polymer morphology was examined by AFM (Nano Wizard II) and FE-SEM (MIRA3 TESCAN). The thermal stability of functionalized polymer was investigated by NETZSCH STA 409 PC/PG under a nitrogen atmosphere (rate of N2 ≈ 1 L h−1). The catalyst copper content was determined by atomic absorption spectrophotometer (GBC Avanta instrument) and EDS (SAME, France) analysis.Monitoring of the reactions and the purity determination of the products were accomplished by TLC on PolyGram SILG/UV 254 silica gel plates.
Synthesis of calix[4]resorcinarene 1
Calix[4]resorcinarene was prepared according to a previously reported procedure.9Synthesis of the 3D-network polymer based on calix[4]resorcinarene 2
The desired polymer was synthesized by adding 42 mmol of formaldehyde to 14 mmol of the prepared calix[4]resorcinarene dissolved in 40 mL NaOH solution (10%) at room temperature. The resultant mixture was heated to 90 °C and maintained at this temperature for 20 h. Next, the excess alkali was washed out of the gel formed with cold water. The gel was allowed to stand at 100 °C for 1 h. Then, the gel was transformed into the acidic form by treatment with the 0.1 M HCl solution. The resulting solid was dried at 100 °C for 10 h.10Synthesis of novel amine-functionalized polymer 3
The synthesized polymer 2 obtained from the above procedure (1.0 g), 3-(triethoxysilyl)propylamine (4.0 g, 18 mmol) were introduced into 50 mL round-bottomed flask containing 10 mL H2O/EtOH at room temperature. The reaction mixture was heated to 90 °C and stirred at this temperature for 20 h. The desired amine-functionalized polymer 3 was collected by filtration and washed several times with hot toluene before being dried at 100 °C.Determination of the amount of amine functionalities grafted onto the polymer
The amount of amine functionalities grafted onto the polymer was determined by elemental analysis to be 3.5 mmol per gram of dry polymer.Preparation of copper(I) iodide salt
To a stirred solution of 10 g copper(II) iodide in 10 mL water was added slowly at room temperature a solution of 7.6 g of anhydrous sodium sulfite, Na2SO3, in 50 mL water. The copper(I) iodide solution first became very dark brown, and then white copper(I) iodide slowly separated. After all the sodium sulfite solution was added and the mixture stirred thoroughly, the copper(I) iodide settled readily and the supernatant liquid was faintly green. The precipitate and supernatant liquid were then poured into about a liter of water to which 1 g of sodium sulfite and 2 mL of concentrated hydrochloric acid were added, and the mixture was stirred well and allowed to stand until all the copper(I) iodide was settled. The supernatant liquid was carefully decanted and the precipitate was quickly washed onto a suction filter (sintered glass type preferred) with dilute sulfurous acid solution. Care should be taken that a layer of liquid covers the salt in the funnel at all times. The copper(I) iodide was then washed four or five times with 20 to 25 mL of glacial acetic acid. During this washing process the suction should be adjusted so that the washed liquid would be sucked through rather slowly. When only a thin film of liquid covered the solid, the next portion of glacial acetic acid was added. The walls of the funnel should be washed each time with the washing liquid. The washed copper(I) iodide with glacial acetic acid was then washed by three 30 mL of absolute alcohol and six 15 mL of anhydrous ether in exactly the same way. After the last portion of ether was removed fairly completely by applying suction for about 30 seconds, the white solid (with filter paper removed) was transferred quickly to a previously dried watch glass and placed in an oven (75 to 100 °C) for 20 to 25 minutes. The sample should be preserved in an airtight bottle. The copper(I) iodide prepared by this method is a white crystalline powder that remains practically unchanged for an indefinite period if kept dry. If all the alcohol is not removed during the washing process, the copper(I) iodide may become slightly discolored on heating or standing. Some samples of anhydrous ether tend to impart a gray tint to the product.11Preparation of novel polymer-supported copper(I) catalyst 4
In a small Schlenk tube, amine-functionalized polymer 3 (1.0 g) was mixed with CuI (0.017 g, 0.1 mmol) in DMF (5 mL). The mixture was stirred for 4 h under a N2 atmosphere at r.t. The solid was isolated by filtration and washed consecutively with acetone (5 mL) and MeOH (5 mL). Finally the resultant product was dried at r.t. for 16 h.Determination of the copper content of catalyst 4
The catalyst copper content was measured using a GBC Avanta, Australia, atomic absorption spectrophotometer (AAS) to be 1.26 percent of the resulted catalyst 4.General procedure for the one-pot synthesis of 1,4-disubstituted 1,2,3-triazoles 7A–F
Benzyl bromide (0.118 mL, 1 mmol) and sodium azide (0.065 g, 1 mmol) were charged into a 25 mL round-bottomed flask containing 3 mL water. Then, phenylacetylene (0.102 g, 1 mmol) and polymer-supported copper catalyst 4 (0.258 g, 0.05 mmol) were added to the reaction mixture. The mixture was heated and stirred under reflux condition till the reaction completion (TLC monitoring). After cooling to r.t., the reaction mixture was vacuum-filtered through a sintered glass funnel and washed with ethyl acetate (5 mL). The resulting solution was poured in a water/ethyl acetate mixture. After extraction of the aqueous phase with ethyl acetate, the combined organic phase was dried over magnesium sulfate and filtered. The resultant solution was concentrated and pure crystalline product was isolated through filtration.Recycling of the polymer-supported copper catalyst 4
After the reaction had been carried out, the mixture was vacuum-filtered onto a sintered-glass funnel. The residue was consecutively washed with CH2Cl2 (5 mL), Et2O (5 mL), EtOH (5 mL), and n-hexane (5 mL). The solid catalyst dried under vacuum after each cycle, and then reused for the next reaction.Results and discussion
This research is essentially focused on developing a novel copper(I)-immobilized catalytic system on the basis of calix[4]resorcinarene that can efficiently modify the CuAAC reaction conditions by combining the advantages of both homogeneous and heterogeneous catalysts.To achieve this objective, the 3D-network polymer 2 was prepared by the reaction of resorcinol and acetaldehyde, followed by the polycondensation of calix[4]resorcinarene 1 with formaldehyde10 as outlined in Scheme 1.
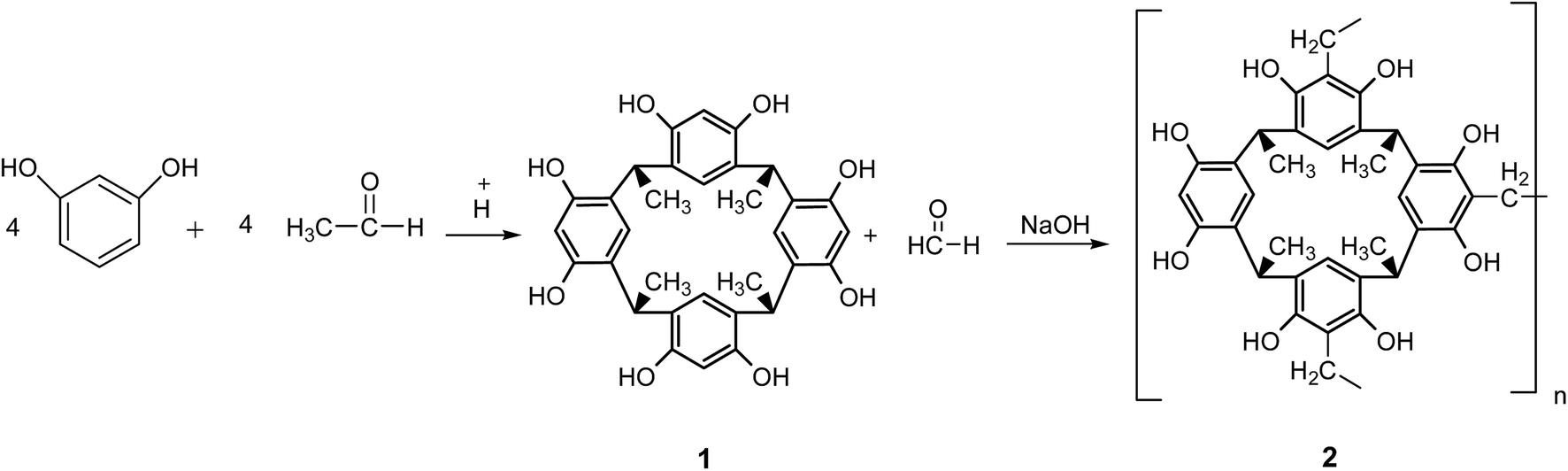 | ||
| Scheme 1 Synthesis of calix[4]resorcinarene 1 and the 3D-network polymer 2. | ||
The structure of compound 1 was established unambiguously from the spectroscopic (IR, 1H NMR, 13C NMR) data (see ESI†). The formation of polymer 2 was also confirmed and characterized by atomic force microscopy (AFM), X-ray diffraction (XRD) and scanning electron microscope (SEM)8a (available in the ESI†).
Among the families of ligands reported for CuAAC reaction, amine groups are prominent as they produce highly robust catalysts capable of conferring outstanding catalytic activity on copper center, often at extremely low catalytic loading. In this regard, we attempted to design, for the first time, a amine functionalized 3D-network polymer based on calix[4]resorcinarene.
To this end, the prepared polymer 2 was treated with 3-(triethoxysilyl)propylamine (APTES) in common solvents to undergo a condensation reaction, yielding amine functionalized polymer 3 in a one-step modification reaction (Scheme 2).
 | ||
| Scheme 2 Synthesis of amine functionalized polymer 3. | ||
The loading amount of the amine moieties on the polymer was determined by elemental analysis. As shown in Table 1, solvent and the amount of added linker (APTES) were optimized. The best result were obtained when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:ethanol mole ratio in the presence of 18 mmol of APTES per gram of polymer was used (Table 1, entry 4). Adding further amount of APTES didn't change the extent of attached amine groups notably (Table 1, entry 5).
:1 water:ethanol mole ratio in the presence of 18 mmol of APTES per gram of polymer was used (Table 1, entry 4). Adding further amount of APTES didn't change the extent of attached amine groups notably (Table 1, entry 5).
| Entry | Solvent | APTES (mmol) | N (wt%) |
|---|---|---|---|
| 1 | Water/ethanol | 8 | 2.32 |
| 2 | Toluene | 8 | 1.03 |
| 3 | Water/ethanol | 15 | 3.82 |
| 4 | Water/ethanol | 18 | 4.95 |
| 5 | Water/ethanol | 20 | 4.97 |
It is noteworthy to highlight that the nitrogen analysis of supported amine 3 (N, 4.95%) indicates that 3.5 mmol of the amine moieties were grafted onto the surface per 1 g of the polymer which show more potential for covalently anchored amine moieties than most previously reported solid-supports.12 Furthermore, the amine polymer 3 is effectively insoluble in various solvents, including water, methanol, ethanol, dimethyl sulfoxide, N,N-dimethylformamide, acetonitrile, dichloromethane, and chloroform. These properties make polymer 3 suitable for being used as an efficient solid-supported reagent or catalyst in various reaction media.
The formation of amine functionalized polymer 3 is of considerable interest since it is the first example of loading of basic groups as well as acidic groups8b on the 3D-network polymer based on calix[4]resorcinarene.
The significant properties of amine functionalized polymer 3 encouraged us to characterize its structure.
IR spectrum of the functionalized polymer 3 is consisted of a broad band in 1120 cm−1 which is representative of Si–O bonds. On the other hand, broad band in 3100–3600 cm−1 revealed that all of the phenolic hydroxyl groups of polymer 2 are not functionalized (see ESI†).
In order to gain an insight into the topography of the surface, AFM was utilized (Fig. 1a and b). All of the AFM scans were taken in ambient air in tapping mode, which is ideal for this kind of polymer.13 More details could be obtained from the 3D height and phase plots (Fig. 1b). Since the amine functionalized polymer 3 shows smaller pores (about 57 nm in diameter, Fig. 2) than polymer 2 (300 nm in diameter, see ESI†), it can be concluded that the propylamine moieties are successfully grafted onto the polymeric backbones.
 | ||
| Fig. 1 AFM images (1.0 μm × 1.0 μm) of amine functionalized polymer 3, (a) 2D and (b) 3D height and phase plots. | ||
 | ||
| Fig. 2 Image profile (the analysis of the height along a linear path) of amine functionalized polymer 3. | ||
Scanning electron microscope of the functionalized polymer 3 was undertaken on a FE-SEM (MIRA3 TESCAN) instrument (Fig. 3). The SEM images confirmed that the average size of pores is in agreement with the AFM data.
 | ||
| Fig. 3 SEM image of amine functionalized polymer 3. | ||
With these results in hand, we believe that 3D-network functionalized polymer 3 could be introduced as a proper porous surface for the inclusion of atomic or molecular guests in chemical reactions.
To investigate the thermal behavior of the functionalized polymer 3 including thermal stability, moisture content and possible decomposition of volatile materials, its thermal gravimetric analysis was carried out under nitrogen atmosphere with a NETZSCH STA 409 PC/PG, from room temperature to 1100 °C, with a heating rate of 10 °C per minute (Fig. 4). According to TG diagram, the weight loss processes of the functionalized polymer could be divided into 3 stages. The first weight loss in the range of 40–120 °C is attributed to the release of physically adsorbed water molecules and organic solvents. A 31.07% weight loss at 200–770 °C is probably corresponding to the thermal decomposition of the attached amine linkers and some parts of polymer which is representative of high thermal stability of this polymeric support. Ultimately the weight loss in the range of 770–1100 °C is associated with decomposition of the polymeric network. Thus, the TGA curve also conveys the clear information that propylamine chains are successfully grafted onto the polymeric backbones.
 | ||
| Fig. 4 TGA profile of amine functionalized polymer 3. | ||
Fortunately, the TGA of polymer 3 demonstrated high thermal stability, with decomposition starting at around 200 °C. The thermal stability of this functionalized polymer allows heat-demanding chemical reactions along with its backbone and side chains without leaching of the active species, thereby providing an excellent means of polymer support and catalyst for conducting chemical reactions.
After the characterization of novel functionalized polymer 3, in the second step, the resultant amine functionalized polymer was further reacted with CuI in DMF to form the corresponding 3D-network polymer-supported Cu(I) catalyst 4 (Scheme 3).
 | ||
| Scheme 3 Synthesis of 3D-network polymer-supported Cu(I) catalyst 4. | ||
The copper content of catalyst, measured by a GBC Avanta, Australia, atomic absorption spectrophotometer (AAS), showed the value of about 1.26 percent of the resulted catalyst (0.2 mmol Cu per gram of the dry polymer-supported catalyst 4).
Energy dispersive X-ray spectroscopy (EDS) was also employed to investigate the elemental composition of the novel catalyst 4 using a SAME, France instrument (Fig. 5).
 | ||
| Fig. 5 EDS analysis of 3D-network polymer-supported Cu(I) catalyst 4. | ||
The copper loadings predicted from EDS analysis (0.62 wt%) does not present a clear trend as we observed in AAS analysis (1.26 wt%). The EDS analysis is capable of measuring copper present on the surface of the catalyst support and cannot detect the copper present inside the pores of the catalyst. So, this absence of the expected trend as in AAS analysis of the copper loading gave us confidence that porous structure of catalyst 4 as well as side-chains have the vital role in supporting of Cu(I) onto the catalyst.
With the copper-incorporated 3D-network polymer 4 in hand, the reactivity of this new type of solid-support transition metal catalyst was investigated. In this regard, owing to the widespread applications of 1,2,3-triazole derivatives, we decided to investigate the synthetic applicability of 4 as a transition metal catalyst for the rapid and efficient construction of these privileged compounds according to the principles of green chemistry.
For this purpose, benzyl bromide, phenylacetylene and sodium azide were chosen as the model substrates to optimize the reaction conditions including the amount of catalyst, solvent and reaction temperature.
Since, water is the most easily available, economical and environment friendly solvent in the word, we first attempted to perform the so-called click reaction in distilled water medium.
To our pleasure, the reaction of model substrates in the presence of catalyst 4 at room temperature in water was successful and the corresponding 1,4-disubstituted 1,2,3-triazole (7A) were obtained in excellent yield without N2 atmosphere. Different solvents and solvent-co-solvent mixtures were tested, and water was found to be the most effective medium for this reaction.
Encouraged by the excellent result, further experiments were designed to survey catalytic virtue of catalyst 4 in the same reaction. As shown in Table 2, the best yield (97%) and the shortest time (5 min) were obtained in the presence of 5 mol% catalyst 4 under reflux conditions (Table 2, entry 3).
| Entry | Catalyst | Catalyst amount/mmol | Temperature/°C | Time/min | Yield% |
|---|---|---|---|---|---|
| 1 | 4 | 0.05 | r.t | 180 | 80 |
| 2 | 4 | 0.05 | 60 | 40 | 86 |
| 3 | 4 | 0.05 | Reflux | 5 | 97 |
| 4 | 4 | 0.025 | Reflux | 180 | 70 |
| 5 | 4 | 0.075 | Reflux | 180 | 82 |
| 6 | — | — | Reflux | 180 | Negligible |
| 7 | CuI | 0.05 | Reflux | 180 | 78 |
To confirm the catalyst efficiency, control experiments were also carried out. Initially, the model reaction was probed in the absence of any catalyst. In this case, only a trace amount of product was detected after 3 h (Table 2, entry 6). Next, the model reaction was examined in the presence of CuI as a homogeneous catalyst and notably decreased yield was resulted (Table 2, entry 7).
To examine the utility and generality of this approach, we have performed the reaction under optimized reaction conditions for the synthesis of a wide variety of 1,2,3-triazole derivatives. Representative results of this synthetic modification are listed in Table 3.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate (5) | Substrate (6) | Product (7) | Time/min | Yield% | Mp/°C |
| A |  |
 |
 |
5 | 97 | 126–128 |
| B |  |
 |
 |
5 | 94 | 127–129 |
| C | 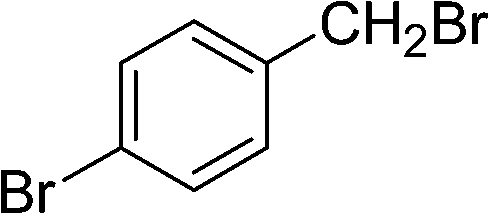 |
 |
 |
10 | 85 | 142–145 |
| D |  |
 |
 |
5 | 88 | 95–97 |
| E |  |
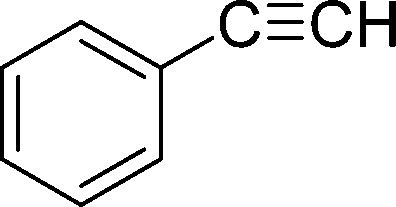 |
 |
30 | 80 | 116–117 |
| F | 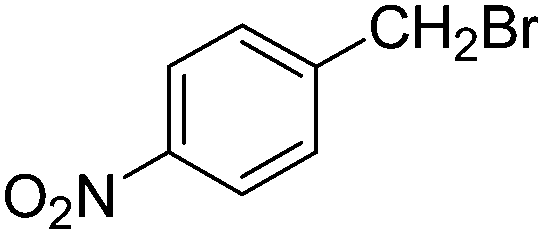 |
 |
 |
5 | 90 | 142–145 |

As it can be observed in Table 3, all the examined benzyl halides with electron donating or electron withdrawing groups in all positions of the phenyl ring (ortho, meta and para) were well compatible with the reaction conditions. Gratifyingly, it was found that the reactions run in air with no need for purification on silica gel.
The products were identified by comparing their physical and spectral data with those of authentic samples.
Considering the above results, we believe that high yields, short reaction times and reactivity of catalyst 4 might be attributed to the existence of a synergistic effect between the cavities of the polymeric support and the immobilized amine groups. The adsorption of substrate into the pores of the functionalized polymer based on hydrophobic interaction between the substrates and polymeric backbone could increase the local concentration of substrates around the active sites of the polymer and effectively promote the reaction.
It is noteworthy that all the reactions proceed without any reducing agent which is commonly used in analogous reactions to prevent Cu(I) from being oxidized to Cu(II). It seems that the porous polymeric matrix could effectively protect the active catalytic sites, copper(I) ions, from interactions leading to oxidation and degradation. On the other hand, the end-functionalized polymer with amine moieties prepares suitable positions for copper ions adsorption to make them stabilized.14
Based on the previous discussion, a possible mechanism for the sequence of events could be expressed such as authentic samples.21 In a first stage, the nucleophilic substitution was carried out on alkyl/aryl halides affording the respective benzyl azides. The amine groups supported on catalyst 4 could function as proton accepters from terminal alkynes activating them to prepare acetylide–copper complex. A newly formed acetylide–copper complex would interact with the azide, activating it toward nucleophilic attack of the acetylide carbon to the ‘end’ nitrogen atom of the azide. Subsequent ring contraction of the generated metallacycle would lead to a copper-triazolide, a direct precursor of the reaction product upon protonation.
It is well known that the reusability of a catalytic system is the key factors that identify whether it has potential applications in industry. Considering this fact, in order to test the catalyst reusability, the reaction of model substrates in the presence of catalyst 4 was carried out under optimal reaction conditions. After completion of each reaction, the catalyst was recovered by simple filtration through a sintered glass-bed, washed with dichloromethane, diethyl ether, ethanol and n-hexane, dried and reused. The results illustrated in Fig. 6 show that the catalyst can be reused for up to five cycles without significant loss in its activity.
 | ||
| Fig. 6 Recyclability of catalyst 4. | ||
Moreover, copper leaching was also determined. AAS analysis of the clear filtrates obtained by filtration after the reaction indicate trace amount of copper ions. This may be rationalized by considering the fact that copper species were properly fixed on the polymeric support. This result indicates that the catalyst was very stable and could endure this reaction's conditions without decomposition or leaching of the active species in the reaction media.
The performances of catalyst 4 and previously reported catalysts were compared in Table 4. Although the methods are effective, the present procedure comparatively affords a truly green process with high yield of the products in shorter reaction times, and carries out these reactions in water without exclusion of air.
| Catalyst | Time (h) | Temperature (°C) | Yield% | Ref. |
|---|---|---|---|---|
| Functionalized chitosan/Cu | 12 | 70 | 99 | 15 |
| Cross-linked polymeric ionic liquid/Cu | 48 | r.t. | 98 | 16 |
| Poly(4-vinyl pyridine)/Cu | 0.4 | 100 | 89 | 17 |
| Nano ferrite–glutathione–copper | 0.17 | 120 MW | 99 | 18 |
| Poly(NIPAM/Im)/Cu | 1.5 | 55 | 99 | 19 |
| Poly(NIPAM/Im)/Cu | 31 | 50 | 94 | 19 |
| MNP–CuBr | 0.3 | 80 MW | 96 | 20 |
| CuNPs/Magsilica | 1 | 70 | 98 | 21 |
| CuNP/activated carbon | 3 | 70 | 98 | 22 |
| A21/CuI | 8 | r.t. | 87 | 23 |
| 4 | 0.08 | 100 | 97 | Present work |
Based on these results, we expect that the catalyst 4 will be a suitable alternative to the existing transition metal catalyst in laboratory and industrial applications.
Conclusion
In summary, we have successfully developed a new type of copper-modified porous organic polymer-based catalyst which combine the catalytic power of transition metal complexes with the architecture of 3D-network polymer and display particularly high efficiency for the Huisgen [3 + 2] cycloaddition. The introduced catalyst can promote the yields and reaction times over 5 repeated runs with very low leaching amounts of supported catalyst into the reaction mixture. The reaction conditions (H2O as solvent, no additive, without exclusion of air) coupled with the sustainability of the catalyst make the described heterogeneous system highly desirable from the point of view of green chemistry. We expect that this new and practical protocol will be useful in academic research and in pharmaceutical development.Acknowledgements
This work was supported by the Research Council at the Shahid Chamran University of Ahvaz.Notes and references
- (a) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS PubMed; (b) R. E. Galian and J. Perez-Prieto, Energy Environ. Sci., 2010, 3, 1488 RSC; (c) A. Corma, Nature, 2009, 461, 182 CrossRef CAS PubMed; (d) D. R. MacFarlane and K. R. Seddon, Aust. J. Chem., 2007, 60, 3 CrossRef CAS; (e) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS PubMed.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589 CrossRef CAS PubMed.
- (a) P. P. Knops-Gerrits, D. E. Devos, F. Thibaultstarzyk and P. A. Jacobs, Nature, 1994, 369, 543 CrossRef CAS PubMed; (b) A.-J. Chen, S.-T. Wong, C.-C. Hwang and C.-Y. Mou, ACS Catal., 2011, 1, 786 CrossRef CAS; (c) L. Wang, X. J. Meng, B. Wang, W. Y. Chi and F.-S. Xiao, Chem. Commun., 2010, 46, 5003 RSC; (d) S. Sahoo, H. Lundberg, M. Eden, N. Ahlsten, W. Wan, X. D. Zou and B. Martin-Matute, ChemCatChem, 2012, 4, 243 CrossRef CAS; (e) S. Farsadpour, L. T. Ghoochany, S. Shylesh, G. Dorr, A. Seifert, S. Ernst and W. R. Thiel, ChemCatChem, 2012, 4, 40 CrossRef; (f) L. Wang, H. Wang, P. Hapala, L. F. Zhu, L. M. Ren, X. J. Meng, J. P. Lewis and F.-S. Xiao, J. Catal., 2011, 281, 30 CrossRef CAS.
- (a) X. H. Liang, S. M. George and A. W. Weimer, Chem. Mater., 2007, 19, 5388 CrossRef CAS; (b) J. Li and Y. Zhang, Chem. Mater., 2007, 19, 2581 CrossRef CAS; (c) Y. Zhang, S. Wei, F. Liu, Y. Du, S. Liu, Y. Ji, T. Yokoi, T. Tatsumi and F.-S. Xiao, Nano Today, 2009, 4, 135 CrossRef CAS; (d) Y. Zhang, J. N. Wang, Y. He, Y. Y. He, B. B. Xu, S. Wei and F.-S. Xiao, Langmuir, 2011, 27, 12585 CrossRef CAS PubMed.
- (a) S. L. Jain, B. S. Rana, B. Singh, A. K. Sinha, A. Bhaumik, M. Nandi and B. Sain, Green Chem., 2010, 12, 374 RSC; (b) S. L. Jain, A. Modak and A. Bhaumik, Green Chem., 2011, 13, 586 RSC; (c) S. Wacharasindhu, S. Bardhan, Z.-K. Wan, K. Tabei and T. S. Mansour, J. Am. Chem. Soc., 2009, 131, 4174 CrossRef CAS PubMed; (d) Y. X. Liu, W. M. Yan, Y. F. Chen, J. L. Petersen and X. D. Shi, Org. Lett., 2008, 10, 5389 CrossRef CAS PubMed; (e) A. R. Katritzky, S. Bobrov, K. Kostyantyn, Y. Ji and P. J. Steel, J. Org. Chem., 2003, 68, 5713 CrossRef CAS PubMed.
- (a) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (b) M. Medal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef PubMed; (c) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef CAS PubMed.
- (a) A. Mouradzadegun and S. Dianat, J. Heterocycl. Chem., 2009, 46, 778 CrossRef CAS; (b) A. Mouradzadegun and F. Abadast, Monatsh. Chem., 2013, 144, 375 CrossRef CAS; (c) A. R. Kiasat, A. Mouradzadegun, S. Elahi and M. Fallah-Mehrjardi, Chin. Chem. Lett., 2010, 21, 146 CrossRef CAS; (d) A. Mouradzadegun and F. Abadast, Tetrahedron Lett., 2013, 54, 2641 CrossRef CAS; (e) A. Mouradzadegun and F. Abadast, Synth. Commun., 2014, 44, 640 CrossRef CAS; (f) A. Mouradzadegun and F. Abadast, Synlett, 2014, 25, 448 CrossRef CAS.
- (a) A. Mouradzadegun, A. R. Kiasat and P. Kazemian Fard, Catal. Commun., 2012, 29, 1 CrossRef CAS; (b) A. Mouradzadegun, S. Elahi and F. Abadast, RSC Adv., 2014, 4, 31239 RSC; (c) A. Mouradzadegun, S. Elahi and F. Abadast, Synthesis, 2015, 47, 630 CrossRef CAS.
- L. M. Tunstad, J. A. Tucker, E. Dalcanale, J. Weiser, J. A. Bryant, J. C. Sherman, R. C. Helgeson, C. B. Knobler and D. J. Cram, J. Org. Chem., 1989, 54, 1305 CrossRef CAS.
- W. H. Awad, J. W. Gilman, M. Nydena, R. H. Harris Jr, T. E. Sutto, J. Callahan, P. C. Trulove, H. C. DeLong and D. M. Fox, Thermochim. Acta, 2004, 409, 3 CrossRef CAS.
- R. Keller and H. Wycoff, Inorg. Synth., 1946, 2, 1 CrossRef.
- (a) M. W. McKittrick and C. W. Jones, Chem. Mater., 2003, 15, 1132 CrossRef CAS; (b) K. Parida, K. G. Mishra and S. K. Dash, Ind. Eng. Chem. Res., 2012, 51, 2235 CrossRef CAS; (c) Y. Zhu, H. Li, Q. Zheng, J. Xu and X. Li, Langmuir, 2012, 28, 7843 CrossRef CAS PubMed.
- S. Magonov and D. Reneker, Annu. Rev. Mater. Sci., 1997, 27, 175 CrossRef CAS.
- S. Díez-González, Catal. Sci. Technol., 2011, 1, 166 Search PubMed.
- M. Chtchigrovsky, A. Primo, P. Gonzalez, K. Molvinger, M. Robitzer, F. Quignard and F. Taran, Angew. Chem., 2009, 121, 6030 CrossRef.
- Y. Wang, J. Liu and C. Xia, Adv. Synth. Catal., 2011, 353, 1534 CrossRef CAS.
- J. Albadi, M. Keshavarz, F. Shirini and M. Vafaie-nezhad, Catal. Commun., 2012, 27, 17 CrossRef CAS.
- (a) R. B. Nasir Baig and R. S. Varma, Green Chem., 2012, 14, 625 RSC; (b) B. Dervauxa and F. E. Du Prez, Chem. Sci., 2012, 3, 959 RSC; (c) A. Megia-Fernandez, M. Ortega-Muñoz, J. Lopez Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Adv. Synth. Catal., 2010, 352, 3306 CrossRef CAS.
- Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 9285 CrossRef CAS PubMed.
- X. Xiong and L. Cai, Catal. Sci. Technol., 2013, 3, 1301 CAS.
- C. S. Radatz, L. d. A. Soares, E. f. R. Vieira, D. Alves, D. Russowsky and P. H. Schneider, New J. Chem., 2014, 38, 1410 RSC.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Adv. Synth. Catal., 2010, 352, 3208 CrossRef CAS.
- M. Keshavarz, N. Iravani, A. Ghaedi, A. Zarei Ahmady, M. Vafaei-Nezhad and S. Karimi, SpringerPlus, 2013, 2, 64 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05234g |
| This journal is © The Royal Society of Chemistry 2016 |