DOI:
10.1039/C6RA05173A
(Paper)
RSC Adv., 2016,
6, 42756-42762
Hybrid mesoporous microspheres from aqueous droplets containing a silica nanoparticle–polymer network in a W/O suspension†
Received
27th February 2016
, Accepted 11th April 2016
First published on 13th April 2016
Abstract
An aqueous dispersion of a reactive copolymer with trimethoxysilyl side chains and silica nanoparticles (SiPs) was added to silicone oil containing 3-aminopropyltrimethoxysilane (APS). The mixture was vigorously stirred for several hours. Consequently, spherical water droplets were formed through the condensation reactions of the copolymer and SiPs as well as APS, which was relocated from the oil phase to the aqueous phase. During the reaction, water was consumed by hydrolysis of the methoxysilyl groups of APS and the droplets gradually solidified. The particulate formation was continuously investigated through optical microscopy. The obtained dried particles were characterized thoroughly using scanning electron microscopy (SEM), field emission scanning electron microscopy (FE-SEM), elemental analysis, 29Si cross-polarization/magic angle spinning nuclear magnetic resonance (29Si CP/MAS NMR) spectroscopy and Brunauer–Emmett–Teller (BET) analysis. The results reveal that these microspheres possess a good compressive strength as APS was successfully incorporated to form Si–O–Si linkages with the copolymer and SiPs to connect the components inside the droplets. The amino functional groups in APS also act to avoid coagulation of the prepared microspheres. Furthermore, it was found that mesoporous silica microspheres embedded with nano-sized silica can also be prepared by calcination of the hybrid microspheres without any fracture. The FE-SEM image of the inner surface of the charred particles shows a uniform distribution of the SiPs and thus confirmed the appropriate morphology of the microspheres.
Introduction
Hybrid microspheres consisting of organic–organic, organic–inorganic or inorganic–inorganic materials have received significant attention because of their potential applications in photonics,1 electronics2 and biotechnology.3 There have been many functional materials used for hybrid microspheres, among these, organically-modified porous silica particles are the most prevalent materials for various applications such as separation media and catalyst supports, because of their unique properties.4–6 In addition, multi-layered silica based microspheres have been constructed and applied in multiple processes.7–9
Various methods10 have been applied for the preparation of silica-based hybrid microspheres: water-in-oil-in-water (W/O/W) double emulsions11 are typically used to fabricate hybrid silica microspheres12,13 to obtain a uniform size and shape. Zhang et al. have revealed a series of preparation of hierarchical porous composites by O/W/O sedimentation polymerization of high internal phase emulsion (HIPE). The obtained beads can be easily handled and used for different applications such as catalysis and separation where large pores are needed to improve mass transport into the pore structure.14 S. H. Kim et al. have already shown a process, consisting of two basic steps, to prepare composite microspheres by simply using mixtures of colloidal materials.15 The preparation of silica microspheres with an encapsulated drug using a single step sonication method16 has also been demonstrated. Mesoporous silica microspheres were first prepared in 1997 (ref. 17 and 18) and have predominantly been used for separation chromatography until now.19,20 Mesoporous silica microspheres with accessible thiol functions21 as well as without any surfactants22 have already been reported. Though a large number of methods have ensued for the preparation of hybrid microspheres of silica nanoparticles (SiPs),23–26 sometimes obtaining a uniform morphology is of great challenge.27,28 Polymer microspheres obtained by templating HIPE, involving no chemical reaction, have been developed by Li et al.29 From O/W emulsion droplets, the oil is slowly removed and the polymer concentrates without any chemical reactions occurring and this eventually leads to solid microspheres.
Recently, we have reported a new class of hybrid hydrogels in which pre-prepared copolymers with reactive side chains are crosslinked using inorganic nanoparticles as multifunctional crosslinking points.30–33 The hybrid hydrogels could be fabricated by simply mixing an aqueous solution of the copolymer with alkoxysilyl side chains and an aqueous suspension of colloidal silica; allowing the resulting mixture to stand for incubation. Such a simple process to form hybrid hydrogels results in desirably shaped hydrogels for use in various techniques. Silica nanoparticles of a smaller size, from 5 to 30 nm, are usually used for the fabrication of hybrid hydrogels. When larger sized silica nanoparticles were used as multiple crosslinkers, the gelation properties became weaker because the number of crosslinking points, compared to smaller particles, was not enough for a network structure connected by the reactive copolymer to be formed. Larger sized silica particles, however, are probably coagulated through inter-particle connections by the reactive copolymers. Microspherical hybrid hydrogels were easily prepared using gel formation in a W/O suspension, and their diameter could be tuned using various parameters such as the composition of the hydrogel and suspension media.33 The preparation of microspheres without a silane coupling agent is challenging because they tend to coalesce or aggregate especially during washing. If a silane coupling reagent containing amino-propyl groups is added to the reaction medium, it can be dissolved in the aqueous media and hydrolysed, and can subsequently form siloxane linkages to connect the silica nanoparticles and reactive copolymer. As a consequence, the pores formed due to condensation of the aqueous phase can be partially filled up. This silane coupling reagent can encapsulate the reactive outer sites to avoid aggregation of the individual particles.34–37 In this article, we demonstrate invention of a new class of hybrid microspheres inspired by the hybrid hydrogels composed of silica nanoparticles and the reactive copolymer described above. To fabricate the hybrid microspheres, herein, silica nanoparticles of 40–50 nm were used in the preparation to avoid gelation. An aqueous mixture of SiPs and a reactive copolymer, poly(N,N-dimethylacrylamide-co-3-methacryloxypropyltrimethoxysilane), was dispersed in silicone oil containing 3-aminopropyltrimethoxysilane (APS) as the suspension medium. In this work, we successfully prepare hybrid microspheres from larger sized silica nanoparticles (40–50 nm). However, silica nanoparticles of a smaller size (14.3 nm) immediately formed a gel with the copolymer, which might be due to the minimum use of APS, rather than using the amount of APS stated in previous literature.33
We confirm that the W/O suspension could be stabilized with APS, which is probably due to electrostatic repulsions of the amine groups at the interface of the aqueous particles.
Experimental
Materials
N,N-Dimethylacrylamide (DMAA) and APS were purchased from Wako Chemicals (Tokyo, Japan). DMAA was used immediately after inhibitor removal. α,α′-Azobisisobutyronitrile (AIBN) was collected from Nacalai Tesque (Kyoto, Japan) and recrystallized from methanol. Colloidal SiPs suspensions with a mean diameter of 8–11 nm and 40–50 nm were bought from Nissan Chemicals (Japan) and the silicone oil was from Shin-Etsu Chemical Co. Ltd. 3-Methacryloxypropyltrimethoxysilane (MAPTS) was collected from TCI (Tokyo, Japan) and all the other reagents were analytical grade and used as received.
Synthesis and analysis of copolymer
The reactive copolymer poly(N,N-dimethylacrylamide-co-3-methacryloxypropyltrimethoxysilane) (pSiDm) was synthesized by free radical copolymerization of N,N-dimethylacrylamide (DMAA) and 3-methacryloxypropyltrimethoxysilane (MAPTS) in methanol at 60 °C. Typically, the required amounts of DMAA (9.91 g; 100 mmol) and MAPTS (0.248 g; 1 mmol), and 49.5 mg of AIBN (0.5 wt% of DMAA) were dissolved in methanol in a three-necked round-bottomed flask under stirring. Oxygen was eliminated by bubbling nitrogen through the solution for 40 min. The flask was then placed in an oil bath and the reaction was carried out for 6 hours. A white product was precipitated out by transferring the solution into a large amount of diethyl ether. The product was successively collected by conducting several times dissolution and re-precipitation in methanol and diethyl ether, respectively, and then dried under vacuum at room temperature. A homopolymer of DMAA, i.e. pDMAA, was also prepared as a reference following the same procedure as above. Fourier transform infrared (FT-IR) spectroscopic analysis of pDMAA and pSiDm was conducted with a JASCO FT/IR-4100 spectrometer (JASCO, Japan). Powdered samples were prepared by dispersing the polymers in KBr and compressing the mixture to form disks. 1H NMR spectra of the dried polymers were recorded using a JEOL JNM-LA400 instrument (JEOL, Tokyo, Japan) at 400 MHz with CDCl3 as the solvent and tetramethylsilane as the internal standard. The number average and weight average molecular weights (Mn and Mw) as well as the polydispersity index (PDI) were determined using size exclusion chromatography (SEC) with a Shodex Asahipak GF-7M HQ Column (Showa Denko K. K. Japan) using dimethylformamide (DMF) containing 0.01 M lithium chloride as the eluent. Polystyrene standards were used for calibration.
Synthesis of hybrid microspheres
To prepare the hybrid microspheres, a 10% pSiDm aqueous solution and 10% SiPs aqueous suspension were prepared separately. An inorganic–organic (I/O) suspension was prepared by mixing them in an equal ratio. Hybrid microspheres were prepared by vigorously stirring this suspension in silicone oil at room temperature. APS was added to the silicone oil as an anticoagulant agent prior to the addition of the aqueous (I/O) suspension. After several hours, the prepared microspheres were collected by removing the supernatant from the top of the mixture, washing the resulting solid several times with toluene and drying it under vacuum.
Calcination of hybrid microspheres
The obtained microspheres were placed in an electric muffle furnace. After calcination at a given temperature of 750 °C for 2 hours (heating rate of 5 °C per minute), porous silica microspheres were obtained.
Characterization of synthesized materials
Optical micrographs were collected with an optical microscope (Olympus CX31, Olympus Co. Japan). A drop of the prepared microspheres in silicone oil was placed on a microscope slide and pictures were obtained randomly from different areas of the same sample. To observe the morphology of the obtained particles, SEM (Carry Scope JCM-5700, JEOL, Tokyo, Japan) and FE-SEM (Hitachi SU8000) were used. Size distribution analyses were performed using all the particles in the SEM image and graphs were produced using AZO V250 software. Elemental analyses were carried out on Micro Corder JM10 apparatus (J-Science Lab, Japan). 29Si CP/MAS NMR spectra were obtained at 25 °C using a Varian Unity Inova AS400 (Varian, California, U.S.A) equipped with a Varian 7 mm VT CP/MAS probe at a static magnetic field of 9.4 T. The surface area with pore size was obtained using a BET method with a NOVA2200e (Quantachrome instruments, USA). The compressive strength measurements of the microspheres were carried out with a tensile and compression tester MCT-211 (Shimadzu Co. Ltd., Japan). The compressive strength was calculated at 20% compression strain and used for comparison.
Results and discussion
Preparation of copolymer
Water-soluble reactive copolymers were synthesized by free radical copolymerization of hydrophilic and reactive side chain-branched monomers. The copolymer, pSiDm, was synthesized by free radical polymerization of DMAA and MAPTS (Scheme S1, ESI†). Table 1 lists the molecular weight and polydispersity of the prepared copolymer, pSiDm, and the homopolymer, pDMAA. The 1H NMR spectrum of the copolymer shows the characteristic signals of DMAA and MAPTS, i.e., δ = 0.64 ppm (–SiCH2) and δ = 3.56 ppm [–Si(OCH3)3] for MAPTS, and δ = 2.9 ppm [6H of the –N(CH3)2 group] for DMAA as shown in Fig. S1 (ESI†). The intensities of the broad peak with a peak maximum at 2.9 ppm and another peak at 0.64 ppm were used to calculate the content of each monomer in the copolymer as shown in Table 1. FT-IR spectroscopic analysis of the polymers indicated the appearance of a new absorption peak at 1720 cm−1 after the copolymerization of DMAA with MAPTS, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups from MAPTS. Both the 1H NMR and FT-IR results confirm that the copolymerization of DMAA and MAPTS has taken place.31,38–40
O groups from MAPTS. Both the 1H NMR and FT-IR results confirm that the copolymerization of DMAA and MAPTS has taken place.31,38–40
Table 1 Molecular weights and the polydispersity index of the prepared polymers
| |
Observed n/ma in pSiDm |
Mw × 105 |
PDI |
| n/m = molar ratio of DMAA and MAPTS from 1H NMR analysis. |
| pSiDm |
101 |
1.34 |
2.77 |
| pDMAA |
— |
1.75 |
3.56 |
Preparation of porous hybrid microspheres and their calcination
The hybrid microspheres were prepared by vigorous stirring of an aqueous suspension of the polymer and SiPs in silicone oil containing APS. Different formulations used for the preparation of the microspheres were named as M1–M5 and are described in Table 2 with the C/N values from elemental analysis. In the different formulations, the amount of APS was different, however the pSiDm and SiPs were kept at a constant weight. In addition to these trial formulations, some other batches with different compositions were also prepared (Table S1, ESI†). But no successful microsphere formation was perceived. It is also worth emphasizing that microsphere preparation in the absence of APS invariably led to complete coagulation (Fig. S2, ESI†). Thus APS was found to be an important contributor during microsphere preparation. No microspheres were observed using the homopolymer and an 8–11 nm SiPs suspension with different percentages of APS (Fig. S3, ESI†). Achieving the porous structure requires removal of the pSiDm; in our case, high temperature calcination was applied to burn off the organic components of the hybrid particles. The resultant silica microspheres maintained a perfect morphology after calcination and only very few broken ones were observed (Fig. S4, ESI†), indicating their thermal stability. In addition, heavily fused silica microspheres were attained (run 7, Table S1, ESI†) using no polymer (Fig. S5, ESI†).
Table 2 Composition of the prepared composite microspheres with elemental analysis and C/N ratio results
| Formulations |
pSiDm aq.a (mL) |
SiPs aq.b (mL) |
APSc (mL) |
Yield (g) |
Elemental analysis |
| N% |
C% |
H% |
C/N |
[pSiDm] = 10 wt%. [SiPs] = 10 wt%. 100 mL of silicone oil (100 CS) was used as the suspension medium. No microspheres were obtained. Calculated for pSiDm copolymerized in 101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Calculated for C3H11NO3Si (three methoxy groups were hydrolysed). :1 ratio. Calculated for C3H11NO3Si (three methoxy groups were hydrolysed). |
| M1d |
5 |
5 |
0 |
|
— |
— |
— |
— |
| M2d |
5 |
5 |
0.1 |
|
— |
— |
— |
— |
| M3 |
5 |
5 |
0.5 |
1.32 |
6.90 |
26.75 |
4.74 |
3.89 |
| M4 |
5 |
5 |
1.0 |
1.60 |
7.51 |
27.07 |
4.98 |
3.61 |
| M5 |
5 |
5 |
2.0 |
1.80 |
8.57 |
27.54 |
5.78 |
3.21 |
| pSiDme |
|
|
|
|
13.79 |
60.3 |
9.06 |
4.37 |
| APSf |
|
|
|
|
7.8 |
40.2 |
9.5 |
2.57 |
Morphological and compositional analysis
During the experiment, the microspheres were collected at regular intervals from the silicone oil to observe them through a polarizing microscope. Fig. 1 displays periodically obtained images of the M3 microspheres, formed in silicone oil after different stirring times. Using the images, for the first several hours, larger emulsion droplets were observed to exist. After several hours of stirring, solid microspheres were obtained. Although initially 90% of the aqueous phase consisted of water, thermogravimetric analysis (TGA, Fig. S6, ESI†) showed a weight loss of 10–12% before reaching 100 °C, which indicates the presence of a negligible amount of water in the prepared microspheres. Besides, the yield (Table 2) was higher in amount than the solid content in the aqueous phase (theoretically, pSiDm and SiPs = 1 g). The above analyses indicate that due to vigorous stirring of the W/O emulsion, the water is slowly removed resulting in the solidification of pSiDm and SiPs with the intrusion of APS in emulsion droplets.29,41 Table 2 also shows the data obtained from elemental analysis. The theoretical C/N value for bare pSiDm is 4.37 and the C/N value periodically decreased upon the addition of APS. After 2 hours of stirring the C/N ratio for the M3 microspheres showed a minute increase to 4.65 (Table S2, ESI†) instead of decreasing, which may be due to the unstable position of the microspheres in the very initial stages of formation. However, with time the C/N value was found to decrease to a constant value (C/N = 3.87–3.89) after 6 hours of stirring (Table S2, ESI†). This indicates the attachment of more APS with time. During suspension particulate formation, APS was relocated from the oil phase to the aqueous phase. Afterwards, the methoxysilyl groups were readily hydrolyzed to form silanol groups. The decreasing of the C/N value from the M3 to M5 microspheres also denotes the introduction of more APS in the microspheres. Most of the network reactions have been taking place within the very first hours of stirring. Both occurrences i.e. the interconnection of the SiPs and pSiDm with the removal of water as well as the coating of the free surface with APS at the same time were revealed from the optical images and elemental analysis of the M3 microspheres.
 |
| | Fig. 1 Optical micrographs for the M3 microspheres after different stirring times in silicone oil at room temperature; scale bars are 200 μm. | |
The size distributions from the SEM photographs of the hybrid microspheres were investigated (Fig. 2). For M3, almost no agglomeration was observed, however with increase of the APS, a little aggregation was observed as the spheres physically adhere together. Here, the M3 microspheres show a wide particle size distribution with a mean diameter of 48.2 μm.
 |
| | Fig. 2 Size distribution of the (a) M3, (b) M4, and (c) M5 microspheres from SEM analysis; scale bars = 100 μm. | |
However, it was also observed that the mean diameter decreases to 27.9 μm and 21.4 μm for M4 and M5, respectively. This agrees well with the following discussion. The reaction rate for these three types of microspheres will not be identical; besides, a slowly evolving reaction means that the microspheres take a longer time to stabilize and hence are probably more likely to aggregate with inter-spherical covalent siloxane bonds.27 The lower amount of APS present in the preparation of the M3 microspheres provides sufficient time for a greater number of covalent bonds to form between the pSiDm and SiPs, until stabilization occurs through complete covering of the surface of the microspheres by the silanol groups of APS. However, the stabilization of the microspheres requires less time in the presence of larger amounts of APS in the oil phase as the APS readily covers the distributed silica particles, and additionally degradation of the organic components leaves larger pores (Fig. 3b and 4). These observations suggest a well-defined arrangement of the polymer and silica particles with the help of APS as well as support our hypothesis of APS intrusion within the hybrid particles. A hypothetical schematic diagram (Scheme 1) is presented to illustrate the formation of the mesoporous microspheres.
 |
| | Fig. 3 FE-SEM images of the M3 microspheres before (a) and after (b) calcination. | |
 |
| | Fig. 4 FE-SEM images of the cross section of a M3 microsphere after calcination at 750 °C. | |
 |
| | Scheme 1 Schematic illustration of the formation of hybrid porous microspheres from aqueous droplets containing a silica nanoparticle–polymer network. | |
29Si CP/MAS NMR spectroscopy was conducted to analyse the chemical structure of the particles before and after calcination. Fig. 5 shows 29Si CP/MAS NMR spectra of the different microspheres, where the signals were observed due to T2, T3 and Q4 units [Tx = RSi(OSi)x(OR′)3−x: x refers to the number of siloxy groups attached, and Qn = Si(OSi)nOH4−n, n ≤ 4]. The relative area percentages of the emerged curves were calculated using a Lorentz function and are summarized in Table 3 as the composition of Tx and Qn units existing in these microspheres. The peaks at −109 ppm and −113 ppm were assigned to Q4 siloxane groups. Furthermore, each microparticle sample before pyrolysis showed a T3 peak at −68 ppm and a T2 peak, as a shoulder, at −59.5 ppm. The latter two signals are due to siloxane groups where the silicon atom is also bound to a carbon atom in an organic group, in addition to being bound to oxygen atoms. With the increase of APS, the relative percentages of Q4 were observed to decrease, which implies that the number of hydroxyl groups decreased to form Si–O–Si bonds. The relative area percentages of T2 and T3 were found to increase, indicating an increase in the formation of new siloxane linkages (Si–O–Si) of aminopropylsilane with the increase of APS. On the other hand; after calcination, no line appeared at −59.6 ppm or −68 ppm, which indicates an absence of aminopropyl groups suggesting complete degradation of the organic parts. Instead, a new peak at −99.5 ppm was detected along with a peak at −109 ppm corresponding to Q3 and Q4, respectively. The relative area percentage of Q3 gradually increased with increase of the APS addition in the microsphere preparation. For all the microspheres, the higher Q4 values obtained after pyrolysis inferred an increase of the interlinked SiO4 tetrahedrons in the interior of the microspheres.41–44
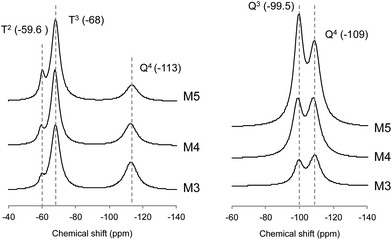 |
| | Fig. 5 29Si CP/MAS NMR spectra of the different microspheres; before calcination (left) and after calcination (right). | |
Table 3 Relative percentages of the different peak areas, derived from 29Si CP/MAS NMR
| |
Before calcination (%) |
After calcination (%) |
| T2 |
T3 |
Q4 |
Q3 |
Q4 |
| M3 |
3.8 |
59.5 |
36.7 |
38.6 |
61.4 |
| M4 |
6.8 |
63.4 |
29.8 |
43.3 |
56.7 |
| M5 |
9.6 |
69.7 |
20.7 |
51.9 |
48.1 |
Surface area and pore size distribution analysis
The specific surface area and pore size of the microspheres, before and after calcination, were achieved directly from BET analysis. Table 4 shows that after calcination, the pore radius and surface area were increased due to the degradation of the organic parts. It is seen that with the increase of the APS amount in the preparation, the specific surface area and pore volume decreased. However, before calcination the pore size remained constant even with the increase of APS. This may be due to the spaces inside the aqueous droplets being filled with the excess amount of APS used in the suspension. APS reacts with the silanol groups on the surface of SiPs as a template. After calcination the pore sizes of the mesoporous particles increase, showing a descending order with the increase of APS. As the organic phase of APS is also burned off from the interspaces of the composite, the microspheres containing a larger amount of APS may provide a larger size of internal pores.
Table 4 Summary of the BET analysis of hybrid microspheres before and after calcination
| |
Specific surface area (m2 g−1) |
Pore radius (nm) |
Pore volumea (cc g−1) |
| Before calcination |
After calcination |
Before calcination |
After calcination |
Before calcination |
After calcination |
| Calculated from BJH desorption. |
| M3 |
30.82 |
112.24 |
1.53 |
7.09 |
0.022 |
0.363 |
| M4 |
29.33 |
86.63 |
1.51 |
6.01 |
0.021 |
0.233 |
| M5 |
22.69 |
77.03 |
1.49 |
4.11 |
0.017 |
0.130 |
Compressive strength analysis
The average compressive strength of the hybrid microspheres, prepared under different preparation conditions, was evaluated from the stress–strain curves at 20% strain and is illustrated in Table 5. The compressive strength analysis was carried out on 5 microparticles of almost similar size for each type. It was observed that the hybrid microspheres have a very high strength in the megapascal range. With the increase of the APS addition the compressive strength was found to be improved, as APS is introduced inside the particles increasing the crosslinking between the polymer and SiPs.33,37 In the present study, the compressive strength was found to be higher than that of previous reports,33 which may be because of the use of 40–50 nm SiPs in the preparation instead of the use of smaller sized SiPs, as smaller sized SiPs can show agglomeration or poor dispersion behaviour.45
Table 5 Compressive strength of the different microspheres
| |
Average diametera (diameter range) (μm) |
Compressive strength at 20% strain (MPa) |
| Five similarly sized particles were analyzed. |
| M3 |
31.5 (30.5–32.5) |
81.7 |
| M4 |
31.45 (27.5–36.5) |
115.5 |
| M5 |
30.03 (28.4–31.5) |
128.5 |
Conclusions
Porous inorganic–organic hybrid microspheres with a crosslink structure of Si–O–Si groups were synthesized by a template preparation using a silica nanoparticle–crosslinked polymer network as the template as well as a coating agent, APS, and a W/O emulsion system. Aminopropyl functional groups were successfully incorporated into the microspheres embedded with alkoxysilyl groups containing the polymer and silica nanoparticles. No evaporation technique was required to remove the solvent in the preparation of the microspheres. FE-SEM analysis of the cross-section proved a homogeneous inside structure of the microspheres. 29Si CP/MAS NMR and elemental analysis confirmed the presence of APS both outside and inside of the microspheres, which consequently showed improved mechanical strength. The size and shape of the microspheres were unchanged upon pyrolysis, even at 750 °C. In general, only very few deformed or cracked structures were observed. BET analysis confirmed a mesoporous structure formation after calcination. Future work will be targeted at controlling the particle morphology and elucidating the influence of the composition and interfacial interactions on the internal pore architecture of the products. These uniform mesoporous particles may be used in multiple applications such as drug release or separation science.
Acknowledgements
This study was supported by the Industrial Technology Research Grant Program from the New Energy and Industrial Technology Development Organization (NEDO) of Japan, and Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Notes and references
- H. Satoh, Y. Saito and H. Yabu, Chem. Commun., 2014, 50, 14786–14789 RSC.
- C. S. Wagner, S. Shehata, K. Henzler, J. Yuan and A. Wittemann, J. Colloid Interface Sci., 2011, 355, 115–123 CrossRef CAS PubMed.
- H. Yang, Y. Liu, Q. Shen, L. Chen, W. You, X. Wang and J. Sheng, J. Mater. Chem., 2012, 22, 24132–24138 RSC.
- K. Nakanishi, R. Takahashi, T. Nagakane, K. Kitayama, N. Koheiya, H. Shikata and N. Soga, J. Sol-Gel Sci. Technol., 2000, 17, 191–210 CrossRef CAS.
- M. Chen, X. Wang, Z. Ye, Y. Zhang, Y. Zhou and W. S. Tan, Biomaterials, 2011, 32, 7532–7542 CrossRef CAS PubMed.
- S. M. Hassan and Y. Kitamoto, Mater. Chem. Phys., 2015, 167, 49–55 CrossRef.
- H. Ji, S. Wang and X. Yang, Polymer, 2009, 50, 133–140 CrossRef CAS.
- M. Ji, H. Liu and X. Yang, Polym. Chem., 2011, 2, 148–156 RSC.
- J. L. Hu, L. B. Luo, X. Z. Yang, R. S. Yao, H. B. Zhang and H. S. Qian, RSC Adv., 2013, 3, 25620–25626 RSC.
- I. V. Melnyk, Y. L. Zub, E. Veron, D. Massiot, T. Cacciaguerra and B. Alonso, J. Mater. Chem., 2008, 18, 1368–1382 RSC.
- B. P. Binks, J. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- J. Zhang, X. Ge, M. Wang, J. Yang, Q. Wu, M. Wu, N. Liu and Z. Jin, Chem. Commun., 2010, 46, 4318–4320 RSC.
- T. M. Choi, J. G. Park, Y. S. Kim, V. N. Manoharan and S. H. Kim, Chem. Mater., 2015, 27, 1014–1020 CrossRef CAS.
- H. Zhang, G. C. Hardy, Y. Z. Khimyak, M. J. Rosseinsky and A. I. Cooper, Chem. Mater., 2004, 16, 4245–4256 CrossRef CAS.
- S. H. Kim, Y. S. Cho, S. J. Jeon, T. H. Eun, G. R. Yi and S. M. Yang, Adv. Mater., 2008, 20, 3268–3273 CrossRef CAS.
- A. Kulak, S. R. Hall and S. Mann, Chem. Commun., 2004,(4), 576–577 RSC.
- M. Gruen, I. lauer and K. K. Unger, Adv. Mater., 1997, 9, 254–257 CrossRef CAS.
- Q. Huo, J. Feng, F. Schueth and G. D. Stucky, Chem. Mater., 1997, 9, 14–17 CrossRef CAS.
- K. K. Unger, D. Kumar, M. Grun, G. Buchel, S. Ludtke, T. Adam, K. Schumacher and S. Renker, J. Chromatogr. A, 2000, 892, 47–55 CrossRef CAS PubMed.
- M. Shahruzzaman, M. Takafuji and H. Ihara, J. Sep. Sci., 2015, 38, 2403–2413 CrossRef CAS PubMed.
- I. V. Melnyk, Y. L. Zub, E. Veron, D. Massiot and T. Cacciaguerra, J. Mater. Chem., 2008, 18, 1368–1382 RSC.
- C. E. Fowler, D. Khushalani and S. Mann, Chem. Commun., 2001,(19), 2028–2029 RSC.
- W. C. Lin, W. Fan, A. Marcellan, D. Hourdet and C. Creton, Macromolecules, 2010, 43, 2554–2563 CrossRef CAS.
- G. Liu, H. Zhang, X. Yang and Y. Wang, Polymer, 2007, 48, 5896–5904 CrossRef CAS.
- G. Liu, L. Li, X. Yang and Z. Dai, Polym. Adv. Technol., 2008, 19, 1922–1930 CrossRef CAS.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Chem. Mater., 2000, 12, 3481–3487 CrossRef CAS.
- L. Gonzalez, M. Baoguang, L. Li, J. H. Hansen, S. Hvilsted and A. L. Skov, Macromol. Mater. Eng., 2014, 299, 729–738 CrossRef CAS.
- G. Perez, E. Erkizia, J. J. Gaitero, I. Kaltzakorta, I. Jimenez and A. Guerrero, Mater. Chem. Phys., 2015, 165, 39–48 CrossRef CAS.
- Z. Li, X. Wei and T. Ngai, Chem. Commun., 2011, 47, 331–333 RSC.
- M. Takafuji, S. Yamada and H. Ihara, Chem. Commun., 2011, 47, 1024–1026 RSC.
- M. A. Alam, M. Takafuji and H. Ihara, J. Colloid Interface Sci., 2013, 405, 109–117 CrossRef PubMed.
- M. A. Alam, M. Takafuji and H. Ihara, Polym. J., 2014, 46, 293–300 CrossRef CAS.
- M. Takafuji, M. A. Alam, H. Goto and H. Ihara, J. Colloid Interface Sci., 2015, 455, 32–38 CrossRef CAS PubMed.
- M. J. Owen, ACS Symp. Ser., 2013, 1154, 47–56 CrossRef CAS.
- Y. Abe, Y. Honda and T. Gunji, Appl. Organomet. Chem., 1998, 12, 749–753 CrossRef CAS.
- R. O. R. Costa and W. L. Vasconcelos, J. Non-Cryst. Solids, 2002, 304, 84–91 CrossRef CAS.
- S. Yue, Z. Zhang, X. Fan, P. Liu and C. Xiao, Int. J. Polym. Anal. Charact., 2015, 20, 285–297 CrossRef CAS.
- M. A. Rodriguez, M. J. Liso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 1999, 34, 3867–3873 CrossRef CAS.
- J. Da and T. E. Hogen-Esch, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 360–373 CrossRef CAS.
- G. Huynh Ba and J. E. McGrath, Polym. Bull., 1980, 2, 837–840 Search PubMed.
- C. M. Boghin, C. I. Cincu, N. N. Marinescu, M. M. Marinescu, M. V. Dimonie, M. Lecca, G. Popescu, C. G. Oprescu, A. Roşanu and M. Lungu, J. Macromol. Sci., Part A: Pure Appl.Chem., 1985, 22, 591–618 CrossRef.
- M. P. Algi and O. Okay, Eur. Polym. J., 2014, 59, 113–121 CrossRef CAS.
- P. Daniels and H. Gies, Phys. Chem. Miner., 1992, 18, 383–388 CrossRef CAS.
- J. Li, J. Ma, T. Jiang, Y. Wang, X. Wen and G. Li, Materials, 2015, 8, 6004–6017 CrossRef.
- S. Haruehansapong, T. Pulngern and S. Chucheepsakul, Construct. Build. Mater., 2014, 50, 471–477 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05173a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 





