
Inhibition of fibrillation of human serum albumin through interaction with chitosan-based biocompatible silver nanoparticles†
Shubhatam Sena,
Suraj Konarb,
Bodhisatwa Dasc,
Amita Pathakb,
Santanu Dharac,
Swagata Dasgupta*b and
Sunando DasGupta*d
aAdvanced Technology Development Centre, Indian Institute of Technology Kharagpur, Kharagpur 721302, India
bDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: swagata@chem.iitkgp.ernet.in; Fax: +91 3222 255303; Tel: +91 3222 283306
cSchool of Medical Science & Technology, Indian Institute of Technology Kharagpur, Kharagpur 721302, India
dDepartment of Chemical Engineering, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: sunando@che.iitkgp.ernet.in; Fax: +91 3222 255303; Tel: +91 3222 283922
First published on 26th April 2016
Abstract
To understand the pharmacokinetics of administered nanomaterials, it is essential to examine the stability and biological activity of proteins by investigating the physicochemical characteristics of the protein–nanoparticle bioconjugate. In this work, the mechanistic detail of the interaction between human serum albumin (HSA) and silver nanoparticles synthesized using nontoxic and biodegradable chitosan as a reducing and stabilizing agent, have been investigated at the nanobio interface. A combination of spectroscopic, calorimetric, and microscopic techniques have been employed to monitor the interaction process. The results illustrate that the chitosan-mediated silver nanoparticles spontaneously bind to HSA without appreciable conformational changes of the protein. Furthermore the potential of the nanoparticles to inhibit the formation of HSA amyloid-like fibrils, in vitro, has been analyzed using thioflavin T fluorescence, circular dichroism, fluorescence microscopy, and transmission electron microscopy. The experimental observations indicate that interactions between HSA and chitosan-based silver nanoparticles have led to appreciable reduction in amyloid fibril formation. Additionally, cytotoxicity and hemolytic assays are performed to ensure the biocompatibility of the nanoparticles within the application limit.
1. Introduction
Nanomaterials having a wide range of applications at the interface of material science and biology are of great interest because of their dimensional resemblance to biological macromolecules. It has been realized that nanoparticles, when exposed to biofluids, interact readily with serum proteins leading to the formation of a biocorona that defines the biological identity of the particle.1 However, the stability and biological activity of proteins may be lost upon interaction with nanoparticles in the biocorona.2 Essentially, to be useful for biomedical applications, the integrity of the structure of the proteins must be maintained on adsorption to the nanoparticle to avoid any unfavorable effects on cellular processes.3 Thus, the physicochemical behavior of an administered material is strongly dependent on its interaction with plasma protein in the blood stream.4Among the metallic nanoparticles, silver nanoparticles have been extensively used for various biological studies like anticancer activity,5 drug delivery,6 biomolecular detection,7 anti-plaque forming efficacy,8 antibacterial activity9 and also their interaction behavior with different proteins.10–14 Despite this rapid development, the applications of the silver nanoparticles are restricted in nanomedicine due to their potential adverse effects.15 Moreover, many of the synthetic methods for the silver nanoparticles involve reducing agents such as citrate, sodium borohydride, ascorbate, polyester etc. that are associated with environmental toxicity or biological hazards.16–18 However, recently there have been growing interests in utilization of nontoxic biopolymers such as chitosan,17 cyclodextrins,19 and starch,20 acting as both reducing and stabilizing agent, for biosynthesis of the silver nanoparticles. Chitosan, a naturally occurring positively charged polysaccharide with exceptional biocompatibility and biodegradability, has been widely used in several biological applications.21–23 Previously the chitosan-mediated silver nanoparticles (SNPs) are reported to exhibit non-genotoxicity and non-cytotoxicity in macrophages at the bactericidal dose and also show antimicrobial and antibiofilm activities.16,18,24 However before practical realization, it is a prerequisite to understand the behavior of the SNPs towards serum protein and therefore one of the motivations for the present study is to analyze the interaction mechanism of chitosan stabilized silver nanoparticles with human serum albumin (HSA). HSA, the major constituent serum protein in blood plasma, has crucial physiological functions including transport and distribution of various exogenous and endogenous compounds in blood, control of blood osmotic pressure, and so forth.25
The fundamental studies on conjugation of HSA with SNPs will provide molecular level information of the phenomena occurring at the protein–nanoparticle interface leading to the better understanding of the influence of the SNPs on protein fibrillation. Fibrillation is related to the misfolding of proteins resulting in the formation of cross β-sheet-rich amyloid fibrils.26 Recently increasing attention is being directed to probe the effects of wide ranges of nanoparticles having different core composition along with diverse surface functionalities that lead to either promotion,27,28 or inhibition,29–36 or depolymerisation in amyloid fibrillation.37,38 HSA is chosen as a model protein for the present study due to its propensity to form amyloid fibrils under modified solution conditions.39 The effects of various external factors such as, temperature, pH, ionic strength, dispersion media, metal ions, sugars etc. have been investigated earlier on the aggregation process of the serum albumin.40–43 However, only limited literature is available on the influence of nanoparticles on fibrillation of HSA.44,45 Recently, effects of differently functionalized MnFe2O4 nanoparticles on fibrillation process of HSA have been studied.46 Nevertheless, the influence of the SNPs, although relevant in the biological applications, on protein fibrillation has so far been unexplored.
To the best of our knowledge, the mechanistic details about both the interaction behaviour of silver nanoparticles, synthesized using a biopolymer, with a serum protein and also the effect of the particles on the amyloid fibrillation of the protein still remain to be elucidated. The present work involves synthesis and characterization of chitosan-based silver nanoparticles followed by probing the biophysical mechanism of interactions between SNPs and HSA using a combination of spectroscopic, calorimetric, and microscopic techniques. Further, the present study also focuses on monitoring the effects of SNPs on the HSA fibrillation process using various biophysical techniques. Additionally, in view of the fact that any material should possess the primary biocompatibility for biological applications, the toxicity of the synthesized SNPs has also been examined by cell viability and hemolytic assay. Thus the present study, carried out systematically, provides useful insight into protein–nanoparticle interaction, consolidating our understanding of the protein amyloid fibrillation with the possible utilization of the chitosan stabilized silver nanoparticles in modulating the protein amyloid self-assembly process.
2. Materials and methods
2.1. Materials
All the chemicals used were of analytical grade and purchased from commercial sources without further purification. Chitosan from crab shells (viscosity > 400 mPa s), human serum albumin (HSA) and thioflavin T (ThT) were purchased from Sigma-Aldrich Company. All solutions were prepared using high quality milli-Q water (pH 6.85 and resistivity 18.2 MΩ cm at 25 °C).2.2. Synthesis and characterization of chitosan-based silver nanoparticles (SNPs)
Chitosan-based silver nanoparticles were synthesized following the process as reported.17 In brief, 4 mL of 52.0 mM AgNO3 and 10 mL of chitosan (6.92 mg mL−1) dissolved in 1% acetic acid solution were mixed and stirred at room temperature for 15 min. The mixture was then transferred to a 12.5 cm × 2 cm cuvette and kept in a water bath for 12 h at 95 °C to complete the reduction process. The final yellowish brown colour of the solution indicated the formation of SNPs. Excess free chitosan was removed through centrifugation at 8000 rpm for 30 min followed by redispersion in milli-Q water (twice). UV-visible spectrum of the sample was recorded on a Shimadzu-2450 spectrophotometer to examine the characteristic surface plasmon resonance (SPR) band. To identify probable interaction between chitosan and silver, Fourier transform infrared (FTIR) spectroscopy was carried out using a Perkin-Elmer Spectrum RX-II (Model no. 73713, USA) within the scan range 4000–400 cm−1. The morphology and size of the particles were obtained by JEM 2100 high resolution transmission electron microscope operating at an accelerating voltage of 200 kV. The size distribution histograms were prepared by measuring diameters of more than 100 particles from several HRTEM images using ImageJ software (version 1.33; National Institutes of Health). The hydrodynamic radius and zeta potential of the sample was measured by a Malvern Nano ZS instrument (Germany).2.3. Sample preparation for HSA–SNP interaction studies and physical characterization of the bioconjugates
The stock solution of HSA was prepared by dissolving HSA in milli-Q water, and its concentration was determined spectrophotometrically using a molar extinction coefficient of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 219 M−1 cm−1 at 280 nm.47 To study the interaction between HSA and SNPs, the HSA–SNP bioconjugates were prepared by mixing the solution of HSA and SNPs in phosphate buffer (10 mM, pH 7.4) followed by incubation of the mixture at room temperature for 6 h.
219 M−1 cm−1 at 280 nm.47 To study the interaction between HSA and SNPs, the HSA–SNP bioconjugates were prepared by mixing the solution of HSA and SNPs in phosphate buffer (10 mM, pH 7.4) followed by incubation of the mixture at room temperature for 6 h.
The UV-visible spectra of HSA (15 μM) in the absence and presence of varying concentrations of SNPs were recorded using an UV-vis-NIR spectrophotometer, Shimadzu-2450 in the scanning range of 200–350 nm. Each spectrum was corrected with respect to the corresponding blank.
Trp fluorescence emission spectra of HSA (15 μM) solutions incubated in the absence and presence of varying concentrations of SNPs was recorded from 305 nm to 500 nm with an excitation wavelength of 295 nm using a Horiba Jobin Yvon Fluoromax-4 spectrofluorimeter. The slit width and integration time were fixed at 2/2 nm and 0.3 s, respectively. Each spectrum acquired was corrected with respect to corresponding blank spectrum. It was also confirmed that SNPs alone did not fluoresce on excitation at 295 nm. To investigate the interaction of chitosan with HSA, as a control, UV-vis and fluorescence spectroscopy have been performed.
In order to study the energetics of interaction between the SNPs and HSA, isothermal titration calorimetry (ITC) was performed using an isothermal calorimeter (iTC200, Microcal, Northampton, MA) at 298 K. The titration involved sequential addition of 0.8 μL aliquots of protein (0.3 mM) (for a total of 27 injections, 0.8 s duration each) at 120 s intervals, with stirring at 310 rpm, into the reaction cell containing SNP solution (20 μM). The data were fitted using Microcal ORIGIN. The heat of HSA dilution in buffer alone was subtracted from the corresponding integrated ITC profile of HSA–SNP titration for each experiment. The reported thermodynamic quantities were the average of two identical experiments.
Zeta potential measurements of SNPs in absence and presence of HSA (15 μM) at pH 7.4, were carried out using a Malvern Nano ZS instrument (Germany).
For microstructural study of the bioconjugates, the morphologies were observed with a transmission electron microscope TECNAI G2 20S-TWIN (Japan) operated at 80 kV. A drop of bioconjugate solution was applied to a carbon-coated cooper grid and left to dry at room temperature for overnight.
Far-UV circular dichroism (CD) spectra were recorded from 190 to 240 nm at a scan speed 50 nm min−1 on a JASCO-810 automatic recording spectrophotometer, using a quartz cuvette of path length 0.1 cm at room temperature. The final concentration of protein was kept at 2 μM. The protein secondary structure content was determined using an online DICHROWEB server.48
2.4. HSA fibrillation experiments
HSA fibrils were prepared by incubating HSA (50 μM) in absence and presence of varying concentration of SNPs at pH 7.4 (20 mM phosphate buffer) in the presence of 60% (v/v) ethanol at 37 °C for 24 h, using standard procedure.49 For each analysis, phosphate buffer of pH 7.4 (20 mM) was used for dilution of samples.The extent of formation of fibrils of HSA in the absence and presence of SNPs was monitored by thioflavin T (ThT) fluorescence. ThT binding assay was performed by withdrawing aliquots from the sets of solutions at different time intervals. After the samples attained room temperature, ThT was added and incubated for five minutes and ThT fluorescence was measured using a Horiba Jobin Yvon Fluoromax 4 spectrofluorimeter. The protein and dye (ThT) concentrations used for measurements were kept at 2 μM and 10 μM, respectively. Excitation and emission wavelengths were at 450 and 482 nm, respectively. Excitation and emission slit widths were 5 nm each with an integration time of 0.3 s. All spectra acquired were corrected with corresponding blank spectra. All the possible blanks, native HSA, SNPs, and native HSA with SNPs separately were checked and found to be not responsive to ThT fluorescence under the experimental condition used. Each experiment was done in triplicate.
To study the secondary structural changes of HSA during fibrillation process, far-UV CD spectra of the solutions were acquired by the method discussed earlier, followed by quantitative estimation of the protein secondary structure content using the online server. Each measurement was done at least three times.
To monitor fibrillar growth of HSA, fluorescence microscopy was performed using a Leica DM 2500 M microscope equipped with a fluorescence attachment. To achieve the required staining for imaging purpose, 10 μL of protein solutions in the absence and presence of SNPs were incubated with 5 μL of 1 mM ThT. Filter cube no. 2 (Leica I3 11, 513, 878, BZ: 01) was used for ThT excitation and emission. The images were obtained with a Leica DFC 310 FX camera attached with the microscope. All images were acquired at 10×/0.25 (N PLAN EPI). For further visualization of the HSA fibrils, bright field image has also been acquired for the same.
For morphological evolution using transmission electron microscopy, HSA fibrillar suspensions, with and without SNPs, were diluted and placed on carbon coated TEM grids. An aqueous solution of uranyl acetate [1% (w/v)] was used for negative staining of the samples. Samples were then air-dried at room temperature and examined in a TECNAI G2 20S-TWIN transmission electron microscope operating at an accelerating voltage of 80 kV.
2.5. Biocompatibility analysis of SNPs
For analysis of cytotoxicity of the SNPs in different concentrations, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay was conducted following the protocol of Mossman T with certain modifications.50 Osteoblast like MG-63 cells (Source NCCS, Pune) were cultured on poly-L-lysine coated tissue culture plate (Nunc) using Dulbecco's modified eagle medium (DMEM) with 10% fetal bovine serum and 1% antibiotic (Hi Media) solution. For understanding the immediate and sub sequential effect of the SNPs, the analysis was conducted after 24 h and 72 h. After incubation with sample, the media was discarded and the plates were washed with phosphate buffered saline. The plates were incubated with MTT (SRL chemicals, Mumbai) solution (5 mg mL−1) for 4 h. This allowed the living cells to form purple formazan crystals which was dissolved in dimethyl sulfoxide (DMSO) and the optical density was measured at 590 nm. The viability was measured in terms of percentile score with respect to control (only DMEM). The experiment was performed in triplicate for statistical significance.The hemolysis assay was conducted to understand the blood compatibility of the SNPs at different concentrations using the protocol reported earlier.51 Briefly, uncoagulated blood (3% citrate) was collected using a 5 mL syringe (Dispovan). The collected blood was mixed with an equal volume of sterile normal saline followed by centrifugation at 1000 rpm to collect red blood cells (RBCs). Different concentrations of SNPs were mixed with 50 μL RBC suspension and the rest by saline to make the final volume of 1 mL and incubated for 60 minutes at 37 °C. Subsequently, the samples were centrifuged at 1000 rpm and the optical densities were measured at 540 nm. Negative and positive controls were taken as normal saline and 1% Triton-X solution respectively. The experiment was performed in triplicate.
3. Results and discussion
3.1. Characterization of the synthesized chitosan-mediated silver nanoparticles
The surface plasmon resonance (SPR) band is shown by the noble metal NPs due to interaction of NPs with incident light leading to collective oscillation of conducting electrons.52 The UV-vis absorption spectrum of the resulting solution shows the characteristic SPR band centred at about 420 nm indicating the formation of chitosan based silver nanoparticle (Fig. S1, see ESI†).17The FTIR spectra of free chitosan molecule and SNPs are presented in Fig. S2 (see ESI†). The strong band at 3300–3500 cm−1 is characteristic N–H stretching vibration.53 The noticeable decrease in transmittance for SNP in this region can be attributed to attachment to silver. It may also be observed that the band at 1579 cm−1 in Fig. S2a,† corresponding to the N–H bending vibration, is shifted to 1558 cm−1 along with a decrease in intensity in Fig. S2b.†53 This again suggests the attachment of chitosan to silver through nitrogen atoms. It may also be noted that the spectrum of SNP is different in shape from that of free chitosan. Therefore it may be assumed that free chitosan is not present in the SNP solution or at least not to the extent that it would affect the present study. Additionally, to exclude the possibility of any effect of ethanol on SNPs, the FTIR spectrum of the sample, obtained after incubating with 60% ethanol for 24 h followed by centrifugation, is acquired. The observed lack of change in the appearance of spectrum from that of SNPs (Fig. S2b†) confirms that 60% ethanol has no effect on SNPs.
Fig. S3 (see ESI†) illustrates the representative HRTEM image of the synthesized SNPs at pH 7.4. The mean particle diameter of SNP is found to be around 30 nm. The selected area electron diffraction (SAED) pattern, shown in inset of Fig. S3a,† indicates face-centred cubic (fcc) crystal structure of SNP.17 The higher average hydrodynamic diameter of SNP (33 nm) obtained from dynamic light scattering (Fig. S4, see ESI†) may be ascribed to the solvation of the particles in aqueous medium.
Surface charge potentials of SNPs at different pH are plotted in Fig. S5 (see ESI†). The high positive zeta potential value of the synthesized SNPs, prepared in acidic medium, indicates the presence of positively charged chitosan molecules. Zeta potential measurement shows that SNPs have isoelectric point of 10.4 and have a positive charge of about 40 mV at pH 7.4. This large positive value also allows us to predict about the long-term stability of the colloidal dispersion and less likely aggregation of particles due to mutual repulsion between them. The absence of aggregation of particles at the experimental pH is also confirmed through absorption spectroscopy. It is observed that there is only slight reduction in SPR intensity when pH of the SNP solution is made 7.4 (Fig. S6, see ESI†). This result along with other previous observations indicates that at the experimental pH of our study, aggregation of particles does not occur.54
3.2. Physical characterization of the HSA–SNP bioconjugates
 | ||
| Fig. 1 (a) Effect of SNPs in the UV-vis behavior of HSA at different concentrations of SNPs (A–F = 0, 5, 10, 40, 80 and 100 μM) (b) Trp fluorescence spectra of HSA and HSA in presence of increasing concentrations of SNPs (A–G = 0, 8, 16, 40, 52, 80 and 100 μM). | ||
The equilibrium for the complex formation between SNPs and HSA can be represented as follows.

| Aobs = (1 − α)C0εHSA1 + αC0εC1 | (1) |
By using the Beer–Lambert law, eqn (1) can be written as follows.
| Aobs = (1 − α)A0 + αAC | (2) |
 | (3) |
Thus eqn (3) can be modified as
 | (4) |
A linear plot is obtained from the graph of 1/(Aobs − A0) versus 1/[SNP] with slope = 1/Kapp (AC − A0) and an intercept = 1/(AC − A0) (graph shown in inset of Fig. 1a). From this plot the value of Kapp is determined to be 1.2 × 103 M−1. The observed increase in absorption maxima along with high binding constant indicates good affinity of HSA to bind with SNP leading to complex formation.
| Fcorr = Fobs × 10(Aex+Aem)/2 | (5) |
It may be noted here that the interaction study between chitosan and HSA, as a control, has also been carried out using UV-vis and fluorescence spectroscopy. However, no noticeable changes are observed in the spectra of HSA, in both studies, after addition of chitosan at the concentration level used in our work. Thus this result precludes the possibility that the observed changes during the interaction study could be due to the interactions between HSA and chitosan.
 | ||
| Fig. 2 ITC profile for the titration of HSA with SNP at 300 K, pH 7.4. Each peak in the upper panel indicates heat flow for each single injection (μcal s−1) as a function of time (minutes) and lower panel shows an integrated heat profile against molar ratio. The solid line represents the best nonlinear least-squares fit. | ||
| Ka (binding constant, M−1) | n (site size) | ΔH (binding enthalpy, kcal mol−1) | ΔS (entropy change, kcal mol−1 K−1) | ΔG (free energy change, kcal mol−1) |
|---|---|---|---|---|
| 1.01 (±0.12) × 103 | 5.26 | −99.56 (±2.05) | −0.306 | −8.372 |
The ITC results are used here to decipher the nature of interactions between SNP and HSA. It has been reported earlier that the negative value of enthalpy is an outcome of the electrostatic interaction, whereas the positive value of entropy is caused by the hydrophobic interaction during the protein–ligand complex formation.61 However, the thermodynamic parameters (ΔH < 0, ΔS < 0), as revealed from the present study, suggests that binding is essentially driven by enthalpy. This indicates that the electrostatic interactions are the predominant driving force in the binding between HSA and SNP, which have negative and positive charge respectively at pH 7.4. The exothermic binding process also indicates the possible formation of noncovalent bonds during intrinsic bond formation or namely protein–particle interaction.62 The unfavorable contribution to the entropy change i.e. the entropy loss may be due to the restriction of conformation freedom of the protein upon binding to nanoparticle.45 The site size value (n), which is the reciprocal of the binding stoichiometry (N), is found to be 5.26, signifies that each nanoparticle (diameter of 30 nm) is surrounded by at least five HSA molecules (approximate dimensions of HSA 9 × 5.5 × 5.5 nm).63 It is previously established that adsorption of the serum albumin over nanoparticle surface occurs preferably by side-on mode than the end-on mode as the side-on mode leads to greater surface area coverage of the protein (49.5 nm2 against 30.2 nm2).63,64 Thus it may be assumed here also that the five HSA molecules adsorb by the side-on mechanism to form a sparse layer on each SNP as suggested by the value of the degree of surface coverage (5.7%), which is calculated following the method as previously described64,65 (see ESI†).
 | ||
| Fig. 3 (a) TEM image of HSA–SNP bioconjugate (scale bar = 200 nm) (b) far-UV CD spectra of HSA in absence and presence of 100 μM SNPs. | ||
3.3. Studies on fibrillation of HSA in presence of chitosan-mediated silver nanoparticles
 | ||
| Fig. 4 (a) Representative ThT fluorescence spectra of HSA solutions in the absence and presence of SNPs of increasing concentrations (A–O = 0 to 150 μM) after 24 h of incubation at 37 °C. (b) SNP-induced inhibition of HSA fibrillation at increased SNP concentration detected by ThT binding study. Error bars represent standard deviation from the mean value estimated from at least three individual measurements. | ||
To determine half-maximal assembly-inhibition concentration (IC50), the observed amyloid fibril forming inhibition ability of SNPs of varying concentration ranging from 5 to 150 μM, at constant HSA concentration (50 μM), is studied by ThT assay. Fig. 4b displays the relative fluorescence intensities (normalized to the fluorescence intensity of the HSA fibrillar solution without SNPs) at different concentrations of SNPs. From the plot, the value of IC50 i.e. the concentration of SNPs, that is required for 50% inhibition of fibril formation, is found to be 71.5 μM.
The more detailed information regarding the fibrillation process can be obtained from ThT fluorescence kinetics study. Fig. 5 shows the temporal evolution of HSA fibril formation measure by means of ThT assay in the absence and presence of SNPs. The time course of fibrillation of HSA shows a typical sigmoidal curve defined by a lag phase, subsequent exponential growth, and a final equilibrium phase. The experimental data are fitted to the following sigmoidal empirical equation to analyze the kinetics of the fibrillation process.67
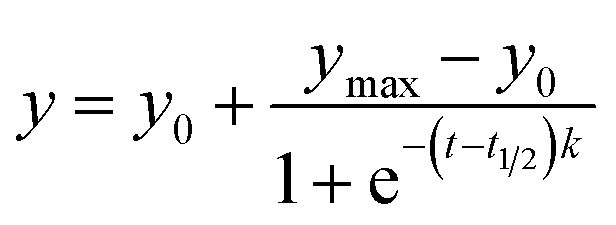 | (6) |
 | ||
| Fig. 5 Fibrillation kinetics of HSA at 37 °C monitored by the temporal development of ThT binding in the absence and presence of SNPs (150 μM). Error bars represent standard deviation from the mean value estimated from at least three individual measurements. | ||
The lag time, which is the time required to form critical nuclei that leads to the formation of fibrils, is given by31
 | (7) |
Values of k, t1/2 and lag time for HSA fibrillation process in absence and presence of SNPs are shown in Table 2. In the absence of SNPs, the main growth of HSA fibrils occurs approximately after 1.1 h (lag time) and attains the plateau region, which corresponds to the final state of the fibrillation process, after ∼18 h. On the other hand, the kinetics of growth of HSA fibrils in presence of SNPs is noticeably delayed with a lag time of ∼4.3 h. Furthermore, slightly lower k value in the presence of the SNPs indicates less efficient fibrillation of HSA. Additionally the significant difference in the values of the plateau for the HSA and HSA in presence of SNPs (Fig. 5) indicates effective inhibition of fibrillation.
| k (h−1) | t1/2 (h) | Lag time (h) | |
|---|---|---|---|
| No particle | 0.32 ± 0.02 | 7.39 ± 0.15 | 1.15 ± 0.40 |
| SNP | 0.27 ± 0.03 | 11.68 ± 0.45 | 4.30 ± 0.60 |
 | ||
| Fig. 6 (a) Far-UV CD spectra and (b) histogram of % secondary structure content of native HSA and HSA fibrillar solution in the absence and presence of 150 μM SNPs. Error bars represent standard deviation from the mean value estimated from at least three individual measurements. | ||
During the formation of fibrils, the native structure of protein is disrupted along with a concomitant increase in β-sheet content. Native HSA is found to contain ∼61% α-helix and only ∼4% β-sheet, whereas after being treated to fibrils, β-sheet content increases to ∼37% and α-helical content decreases to ∼11%. In contrast, presence of SNPs results in ∼67% relative decrease in β-sheet content of along with ∼73% relative increase in α-helix, both with respect to HSA fibrillar solution without SNPs. This observation reveals that SNPs retard the unfolding of HSA and thus preventing the association of HSA monomers to form fibrils effectively, consistent with the results from ThT fluorescence spectroscopy.
 | ||
| Fig. 7 (a) Fluorescence microscopic images (scale bar = 200 μm) and (b) TEM images (scale bar = 200 nm) of HSA solutions in the absence and presence of SNPs (150 μM) after incubation for 24 h at 37 °C in the presence of 60% (v/v) ethanol at pH 7.4: (1) HSA fibrils (2) HSA–SNP. | ||
To further elucidate morphology of HSA fibrillar solution with and without SNPs, TEM study is performed. In case of the HSA fibrillar solution without SNPs (Fig. 7b1), the dense fibrillar structures with complex branching, similar to that reported earlier, are observed43 (for further visualization, bright field image of HSA fibrillar solution in absence of SNPs showing distinct fibrillar growth is presented in Fig. S7, ESI†). On the other hand, presence of SNPs results in the formation of amorphous and globular aggregates (Fig. 7b2). Thus, the microscopic studies, in accordance with other experiments, clearly delineate the role of SNPs towards inhibition of fibrillation of HSA.
As is evident from interaction studies, SNPs form stable bioconjugates with HSA, with negative free binding energy change making the interaction process spontaneous. The electrostatic interaction between HSA and SNPs, as obtained from ITC study and zeta potential measurement, plays the dominant role for the binding process. At pH 7.4, positively charged SNPs (pI 10.4) are able to interact favorably with the negatively charged HSA (pI 4.9)68 through electrostatic interactions which in turn brings the HSA molecules and the nanoparticles close to each other. The results from the various biophysical techniques also indicate that native structure of HSA is retained upon binding to SNPs. We speculate that the stabilization brought about by strong interactions between SNPs and HSA disturbs protein self-association, which in turn leads to reduced formation of HSA amyloid-like fibrils.
Fibrillation process follows a nucleation-growth pattern where the formation of critical nuclei is the key rate-determining step, as it is a thermodynamically unfavored step during fibrillation.35 Strong interactions between the protein and nanoparticles at the early stage of fibrillation affect to form the nucleation species by depletion of active monomers from solution. The inhibition of fibrillation by the reduction in free monomer concentration by nanoparticles has also been reported earlier e.g. inhibition of Aβ peptide fibrillation by thioglycolic acid-stabilized CdTe nanoparticles30 or fibrillation of insulin by carbon dots.35 Here as is evident from ThT fluorescence kinetic study, the addition of SNPs slows down the aggregation process of HSA by a marked extension of the lag phase which is believed to be the activation time required for the formation of critical nuclei. This observation indicates that HSA fibrillation process is initiated much later in presence of SNPs. Thus SNPs may be considered as an efficient inhibitor to retard HSA fibrillation by prolonging the lag time for HSA nucleation, which is supported by the interaction study showing that the nanoparticles bind strongly the proteins to form complex. Furthermore, the significant reduction in the extent of fibrillization, as observed by the equilibrium plateau value at the end of the fibrillation process, indicates lowering of the elongation rate by disturbing the self-assembly pathway.
3.4. Biocompatibility of SNPs
 | ||
| Fig. 8 MTT assay results at different concentrations of SNPs after (a) 24 h (b) 72 h incubation. Error bars represent standard deviation from the mean value estimated from at least three individual measurements. | ||
The hemolysis assay results of SNPs at different concentrations along with positive (Triton-X) and negative (saline) control are presented in Fig. 9. The positive control and negative control represent 100% and 0% hemolysis, respectively. The percentage of hemolysis is found to be 5.3% at 100 μM SNPs, the maximum working concentration for the interaction study, suggesting low hemolytic activity of the nanoparticles. When concentration of the SNPs is increased to 150 μM, the highest concentration used for the fibrillation study, only 6.7% hemolysis is observed. Further with increasing the concentration of SNPs to 300 μM, which is much higher than the application limit, the percentage hemolysis is found to be only 14.4%. Therefore, the chitosan-mediated silver nanoparticles may be considered as hemocompatible in nature and the MTT assay along with the hemolysis assay shows the biocompatibility of the chitosan-mediated silver nanoparticles within the application limit.
 | ||
| Fig. 9 Hemolytic assay of the SNPs at various concentrations (a) 100 μM (b) 150 μM (c) 300 μM. (d) and (e) represent positive and negative control respectively. Error bars represent standard deviation from the mean value estimated from at least three individual measurements. | ||
4. Conclusions
In summary, silver nanoparticles have been successfully synthesized using biodegradable and nontoxic chitosan as a reducing and stabilizing agent and characterized by various techniques. Herein, the influence of the silver nanoparticles on the binding with human serum albumin as well as on the amyloid fibrillation of the protein has been studied comprehensively. The interaction process is monitored using various experimental methods. The apparent association constant (1.2 × 103 M−1) is evaluated from the absorption spectral changes of HSA in presence of SNPs using the Benesi–Hildebrand equation. From the steady state Trp fluorescence spectroscopy, metal-enhanced fluorescence phenomenon is found to be associated with the binding process. ITC study reveals that the formation of SNP–HSA bioconjugate is a thermodynamically spontaneous process, with a negative free binding energy change of −8.372 kcal mol−1, mainly driven by electrostatic interactions-which is also supported by zeta potential measurements. The results indicate binding induces indiscernible conformational changes of HSA. Further the inhibiting effectiveness of SNPs towards fibrillation of HSA is monitored by various biophysical techniques. The ThT binding study shows ∼85% decrease in fluorescence intensity at 150 μM concentration of SNPs indicating reduced amyloid fibril formation. The CD results confirm a relative percentage decrease in the β-sheet content in presence of SNPs to be ∼67% as compared to the HSA fibrillar solution. The formation of amorphous and globular aggregates in the presence of SNPs, as revealed by the microscopic images, also indicates that SNPs inhibits fibrillation of HSA. The ThT kinetic study suggests that the positive interaction of SNPs with HSA leads to the retardation of amyloid-like fibril formation by affecting the nucleation step as well as the growth phase. Moreover, the silver nanoparticles are found to possess good cytocompatibility as well as hemocompatibility within the application limit. Although detailed ‘real’ studies under in vivo conditions are necessary for practical realization, the ability of the chitosan-modified nanoparticles to inhibit fibrillation of the serum albumin indicates of its possible utilization in modulating the protein amyloid self-assembly pathway.Acknowledgements
The authors gratefully acknowledge the financial support provided by the Indian Institute of Technology Kharagpur, India [Sanction Letter no. IIT/SRIC/ATDC/CEM/2013-14/118, dated 19.12.2013] and the use of its Central Research Facility.References
- C. Gunawan, M. Lim, C. P. Marquis and R. Amal, J. Mater. Chem. B, 2014, 2, 2060–2083 RSC.
- M. J. Hajipour, S. Laurent, A. Aghaie, F. Rezaee and M. Mahmoudi, Biomater. Sci., 2014, 2, 1210–1221 RSC.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- V. S. Jisha, K. T. Arun, M. Hariharan and D. Ramaiah, J. Am. Chem. Soc., 2006, 128, 6024–6025 CrossRef CAS PubMed.
- J. Lin, Z. H. Huang, H. Wu, W. Zhou, P. P. Jin, P. F. Wei, Y. J. Zhang, F. Zheng, J. Q. Zhang, J. Xu, Y. Hu, Y. H. Wang, Y. J. Li, N. Gu and L. P. Wen, Autophagy, 2014, 10, 2006–2020 CrossRef PubMed.
- P. K. Brown, A. T. Qureshi, A. N. Moll, D. J. Hayes and W. T. Monroe, ACS Nano, 2013, 7, 2948–2959 CrossRef CAS PubMed.
- S. Paul, D. Paul, G. R. Fern and A. K. Ray, J. R. Soc. Interface, 2011, 8, 1204–1211 CrossRef CAS PubMed.
- J. V. Rogers, C. V. Parkinson, Y. W. Choi, J. L. Speshock and S. M. Hussain, Nanoscale Res. Lett., 2008, 3, 129–133 CrossRef.
- G. Franci, A. Falanga, S. Galdiero, L. Palomba, M. Rai, G. Morelli and M. Galdiero, Molecules, 2015, 20, 8856–8874 CrossRef CAS PubMed.
- V. Banerjee and K. P. Das, Colloids Surf., B, 2013, 111, 71–79 CrossRef CAS PubMed.
- A. Ravindran, A. Singh, A. M. Raichur, N. Chandrasekaran and A. Mukherjee, Colloids Surf., B, 2010, 76, 32–37 CrossRef CAS PubMed.
- J. T. Tai, C. S. Lai, H. C. Ho, Y. S. Yeh, H. F. Wang, R. M. Ho and D. H. Tsai, Langmuir, 2014, 30, 12755–12764 CrossRef CAS PubMed.
- A. Sasidharan, J. E. Riviere and N. A. Monteiro-Riviere, J. Mater. Chem. B, 2015, 3, 2075–2082 RSC.
- V. P. Brahmkhatri, K. Chandra, A. Dubey and H. S. Atreya, Nanoscale, 2015, 7, 12921–12931 RSC.
- S. J. Yu, Y. G. Yin and J. F. Liu, Environ. Sci.: Processes Impacts, 2013, 15, 78–92 CAS.
- D. W. Wei, W. Y. Sun, W. P. Qian, Y. Z. Ye and X. Y. Ma, Carbohydr. Res., 2009, 344, 2375–2382 CrossRef CAS PubMed.
- D. Wei and W. Qian, Colloids Surf., B, 2008, 62, 136–142 CrossRef CAS PubMed.
- P. Jena, S. Mohanty, R. Mallick, B. Jacob and A. Sonawane, Int. J. Nanomed., 2012, 7, 1805–1818 CAS.
- S. Jaiswal, B. Duffy, A. K. Jaiswal, N. Stobie and P. McHale, Int. J. Antimicrob. Agents, 2010, 36, 280–283 CrossRef CAS PubMed.
- M. Valodkar, A. Bhadoria, J. Pohnerkar, M. Mohan and S. Thakore, Carbohydr. Res., 2010, 345, 1767–1773 CrossRef CAS PubMed.
- H. M. Yi, L. Q. Wu, W. E. Bentley, R. Ghodssi, G. W. Rubloff, J. N. Culver and G. F. Payne, Biomacromolecules, 2005, 6, 2881–2894 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- W. S. Xia, P. Liu, J. L. Zhang and J. Chen, Food Hydrocolloids, 2011, 25, 170–179 CrossRef CAS.
- M. Venkatesham, D. Ayodhya, A. Madhusudhan, N. Veera Babu and G. Veerabhadram, Appl. Nanosci., 2014, 4, 113–119 CrossRef CAS.
- V. N. Uversky, N. V. Narizhneva, T. V. Ivanova and A. Y. Tomashevski, Biochemistry, 1997, 36, 13638–13645 CrossRef CAS PubMed.
- E. Gazit, FEBS J., 2005, 272, 5971–5978 CrossRef CAS PubMed.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- W. H. Wu, X. Sun, Y. P. Yu, J. Hu, L. Zhao, Q. Liu, Y. F. Zhao and Y. M. Li, Biochem. Biophys. Res. Commun., 2008, 373, 315–318 CrossRef CAS PubMed.
- A. Das, A. Chakrabarti and P. K. Das, RSC Adv., 2015, 5, 38558–38570 RSC.
- S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S. H. Jeong, A. Ramamoorthy and N. A. Kotov, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef CAS PubMed.
- C. Cabaleiro-Lago, I. Lynch, K. A. Dawson and S. Linse, Langmuir, 2010, 26, 3453–3461 CrossRef CAS PubMed.
- K. Dubey, B. G. Anand, R. Badhwar, G. Bagler, P. N. Navya, H. K. Daima and K. Kar, Amino Acids, 2015, 47, 2551–2560 CrossRef CAS PubMed.
- M. Ansari, M. Habibi-Rezaei, S. Salahshour-Kordestani, A. A. M. Movahedi and N. Poursasan, Protein Pept. Lett., 2015, 22, 594–600 CrossRef CAS PubMed.
- N. Taebnia, D. Morshedi, M. Doostkam, S. Yaghmaei, F. Aliakbari, G. Singh and A. Arpanaei, RSC Adv., 2015, 5, 60966–60974 RSC.
- S. H. Li, L. Y. Wang, C. C. Chusuei, V. M. Suarez, P. L. Blackwelder, M. Micic, J. Orbulescu and R. M. Leblanc, Chem. Mater., 2015, 27, 1764–1771 CrossRef CAS.
- X. B. Zhou, J. Sun, T. T. Yin, F. L. Le, L. C. Yang, Y. N. Liu and J. Liu, J. Mater. Chem. B, 2015, 3, 7764–7774 RSC.
- K. Siposova, M. Kubovcikova, Z. Bednarikova, M. Koneracka, V. Zavisova, A. Antosova, P. Kopcansky, Z. Daxnerova and Z. Gazova, Nanotechnology, 2012, 23, 055101 CrossRef PubMed.
- R. C. Triulzi, Q. Dai, J. H. Zou, R. M. Leblanc, Q. Gu, J. Orbulescu and Q. Huo, Colloids Surf., B, 2008, 63, 200–208 CrossRef CAS PubMed.
- J. Juarez, P. Taboada and V. Mosquera, Biophys. J., 2009, 96, 2353–2370 CrossRef CAS PubMed.
- J. Juarez, M. Alatorre-Meda, A. Cambon, A. Topete, S. Barbosa, P. Taboada and V. Mosquera, Soft Matter, 2012, 8, 3608–3619 RSC.
- A. Stirpe, M. Pantusa, B. Rizzuti, L. Sportelli, R. Bartucci and R. Guzzi, Int. J. Biol. Macromol., 2011, 49, 337–342 CrossRef CAS PubMed.
- N. K. Pandey, S. Ghosh and S. Dasgupta, Int. J. Biol. Macromol., 2013, 61, 424–432 CrossRef CAS PubMed.
- N. K. Pandey, S. Ghosh, D. R. Tripathy and S. Dasgupta, Protein Pept. Lett., 2015, 22, 112–118 CrossRef CAS PubMed.
- C. H. Vannoy and R. M. Leblanc, J. Phys. Chem. B, 2010, 114, 10881–10888 CrossRef CAS PubMed.
- S. Goy-Lopez, J. Juarez, M. Alatorre-Meda, E. Casals, V. F. Puntes, P. Taboada and V. Mosquera, Langmuir, 2012, 28, 9113–9126 CrossRef CAS PubMed.
- S. Sen, S. Konar, A. Pathak, S. Dasgupta and S. DasGupta, J. Phys. Chem. B, 2014, 118, 11667–11676 CrossRef CAS PubMed.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, W668–W673 CrossRef CAS PubMed.
- N. K. Pandey, S. Ghosh and S. Dasgupta, Int. J. Biol. Macromol., 2013, 59, 39–45 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- B. Das, P. Dadhich, P. Pal, P. K. Srivas, K. Bankoti and S. Dhara, J. Mater. Chem. B, 2014, 2, 6839–6847 RSC.
- A. Moores and F. Goettmann, New J. Chem., 2006, 30, 1121–1132 RSC.
- L. Jin and R. B. Bai, Langmuir, 2002, 18, 9765–9770 CrossRef CAS.
- R. K. Saini, A. K. Srivastava, P. K. Gupta and K. Das, Chem. Phys. Lett., 2011, 511, 326–330 CrossRef CAS.
- A. Manivel and S. Anandan, Colloids Surf., A, 2012, 395, 38–45 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- B. J. Yang, R. T. Liu, X. P. Hao, Y. Z. Wu and J. Du, Biol. Trace Elem. Res., 2013, 155, 150–158 CrossRef CAS PubMed.
- C. X. Sun, X. Wu, H. H. Ding, L. L. Zhao, F. Wang, J. H. Yang and X. Y. Liu, J. Fluoresc., 2009, 19, 111–117 CrossRef CAS PubMed.
- J. R. Lakowicz, B. Shen, Z. Gryczynski, S. D'Auria and I. Gryczynski, Biochem. Biophys. Res. Commun., 2001, 286, 875–879 CrossRef CAS PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed.
- M. De, C. C. You, S. Srivastava and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 10747–10753 CrossRef CAS PubMed.
- K. Rezwan, L. P. Meier, M. Rezwan, J. Voros, M. Textor and L. J. Gauckler, Langmuir, 2004, 20, 10055–10061 CrossRef CAS PubMed.
- S. Chakraborti, P. Joshi, D. Chakravarty, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2012, 28, 11142–11152 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. R. Nilsson, Methods, 2004, 34, 151–160 CrossRef CAS PubMed.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS PubMed.
- H. Wiig, O. Tenstad and J. L. Bert, J. Physiol., 2005, 569, 631–641 CrossRef CAS PubMed.
- S. K. R. Chowdhury, A. Mishra, B. Pradhan and D. Saha, Wear, 2004, 256, 1026–1036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05129d |
| This journal is © The Royal Society of Chemistry 2016 |