
Synthesis of core/shell structured Pd3Au@Pt/C with enhanced electrocatalytic activity by regioselective atomic layer deposition combined with a wet chemical method†
Abstract
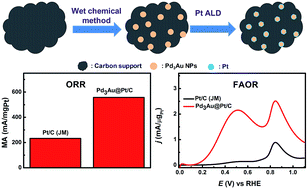
We report the synthesis of Pd3Au@Pt/C by atomic layer deposition (ALD) combined with a wet chemical method. Initially, nearly uniform Pd3Au nanoparticles (NPs) (∼4.7 nm) supported on carbon with a loading of ∼20 wt% were prepared by reducing metal complexes confined in reverse micelles adsorbed on carbon. Next, a Pt thin layer (less than 1 nm) was selectively deposited on purified Pd3Au particles instead of on carbon by regioselective ALD that likely arises from the coverage of nucleation sites on carbon by surfactant molecules in conjunction with catalytic decomposition of the Pt precursor on Pd3Au. The resulting core–shell structured material was subject to TEM, HAADF-STEM/EDX, XRD, XPS and electrochemical measurements. Interestingly, Pd3Au@Pt/C demonstrates a significantly improved electrocatalytic activity toward both formic acid oxidation reaction (FAOR) and oxygen reduction reaction (ORR) compared with Pd3Au/C and state-of-the art Pt/C. This study opens up a unique avenue for the synthesis core/shell structured materials with potential applications in catalysis, electrocatalysis, and so on.
 Please wait while we load your content...
Please wait while we load your content...

