
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of novel analogues of batracylin with synthetic amino acids and adenosine: an unexpected effect on centromere segregation in tumor cells through a dual inhibition of topoisomerase IIα and Aurora B†
Wioleta
Januchta‡
a,
Marcin
Serocki‡
b,
Krystyna
Dzierzbicka
*a,
Grzegorz
Cholewinski
a,
Monika
Gensicka
a and
Andrzej
Skladanowski
b
aDepartment of Organic Chemistry, Chemical Faculty, Gdansk University of Technology, G. Narutowicza 11/12, 80-233 Gdansk, Poland. E-mail: krydzier@pg.gda.pl
bDepartment of Pharmaceutical Technology and Biochemistry, Faculty of Chemistry, Gdansk University of Technology, 11/12 G. Narutowicza St, 80-233 Gdansk, Poland
First published on 26th April 2016
Abstract
In the search for new anticancer agents we designed and synthesized batracylin derivatives with linking synthetic amino acid side chains of different lengths and adenosine. Unexpectedly, we have found that in water and the culture media adenosine–amino acid–BAT conjugates form supramolecular structures and this prevents these compounds from entering cells. Consequently, these compounds exerted no biological activity when tested towards two human cell lines, lung adenocarcinoma (A549) and human leukemia (HL-60). In contrast, several amino acid–BAT precursors showed up to 25-fold enhanced cytotoxic activity compared to BAT and these compounds strongly interfered with DNA topoisomerase II activity and its cellular functions. In particular, these conjugates inhibited centromere segregation during mitosis in drug-treated tumor cells by preventing topoisomerase II-dependent Aurora B activation.
Introduction
Medicinal chemists continue to develop novel cytotoxic agents with new and unique mechanisms of action, however, many of these compounds still lack tumor selectivity or have not been therapeutically useful due to undesired toxic effects or low bioavailability in vivo. Apart from this classical approach to design and synthesize new molecules with antitumor activity, there is yet another line of activity in medicinal chemistry to combine different molecules with defined functions and activities to produce the so-called ‘functionalized’ molecules or drugs. In this approach, one can conjugate cytotoxic drugs with other molecules such as e.g. monoclonal antibodies which bind to specific cellular markers on the surface of tumor cells and offer an alternative therapy that is tumor-specific and/or less toxic. This direction has recently attracted a great deal of attention (for review see Chari 2008).1 Another example can be to synthesize ‘functionalized’ cytotoxic molecules by combination of with other small molecules, such as proteins, peptides or sugars.Batracylin (8-aminoisoindolo[1,2-b]quinazolin-12(10H)-one, BAT) exhibits excellent antitumor properties2–5 and inhibits the catalytic activity of DNA topoisomerases that leads to production of DNA breaks and induction of the unscheduled DNA synthesis (UDS) in nonproliferating cells.6,7 At the same time, in pre-clinical studies BAT produced severe toxicity in rats and mice. In addition, even between rodents different toxicity of BAT was observed and oral administration batracylin to rats was found to significantly more toxic, compared to mice. The BAT toxicity observed across all studied species was caused by presence of its toxic metabolite, N-acetyl-batracylin (ABAT), following metabolism mediated by N-acetyl-transferase 2 (NAT2).8–10 First-in-human study was conducted in pharmacogenetically selected thirty-one patients with advanced refractory solid tumors and lymphomas and a slow acetylator NAT2 genotype.11 The objectives included determination of the safety, tolerability, maximum tolerated dose (MTD) and pharmacokinetics (PK) of batracylin and its metabolites. Although no objective responses were observed, even at doses of BAT as high as 400 mg per day for 7 days (in a 28 day cycles), patients tolerated doses which were about 20-fold higher than the MTD in rats and 70% of the MTD in mice.
The low water solubility of BAT, its high toxicity as well as large doses required for anticancer activity, are the causes that drawn attention to the synthesis of its new analogues.12,13 The synthesis of different types of BAT derivatives was described in the 1990s.14–16 The new compounds were prepared by reacting the batracylin with natural amino acids or short peptides containing additional amino or carboxyl groups. These derivatives were sufficiently soluble in water and reconverted into batracylin by the action of enzymes like peptidases which are present in blood plasma and other organs. Accordingly, these derivatives have been shown to exhibit the cytotoxic activity that is comparable to that of batracylin.14,15 In 1992, the synthesis of β-L-fucose derivative of batracylin was published.16 The linkage of β-L-fucose derivatives to peptide conjugates of batracylin led to an active compound. The procedure was also reported of the synthesis of batracylin analogues modified by carbonylation and based on the use 1,2-dibromobenzenes and 2-aminobenzyl amine as substrates and palladium as the catalyst.17 The desired products were isolated in moderate to good yields (36–80%) with the installation of two molecules of carbon monoxide. The synthesis of N-sulfonoamido analogues was also presented, which contain alkyl and aromatic groups on the batracylin pharmacophore in the presence of pyridine in [b-3C-im][NTf2] ionic liquid and were further assayed for biological activity. Some of synthesized compounds exhibited potent inhibitory activity against human type I DNA topoisomerase.18
We propose that yet another group of the compounds that can be conjugated with BAT is adenosine (6-amino-9-β-D-ribofuranosyl-9-H-purine, ADO), which is an endogenous purine nucleoside. Adenosine is released from cells or is formed outside the cell in the organism, and binds specifically to its receptors, which are present on the cellular membrane.19,20 There are four types of adenosine receptors: A1, A2A, A2B and A3, which belong to the G-protein-coupled family of receptors and frequently have opposing effects on cell signaling pathways.21,22 For example, A1 receptors stimulate formation of giant multinucleated cells from monocytes, whereas A2 receptors inhibit this process. Similarly, A2A adenosine receptors are generally anti-inflammatory, whereas A2B and A3 receptors are implicated in proinflammatory action of adenosine.23 Even more importantly, adenosine by its binding to membrane receptors can induce cell death by apoptosis. The molecular mechanism of this pro-apoptotic activity of adenosine involves modulation gene expression of genes such as Bax, Bad, and Puma, that leads to disruption of the mitochondrial transmembrane potential and activation of caspase-9 and caspase-3.24 In this way, combined effect of cytotoxic drug and adenosine can increase cell killing effect induced by the conjugate. However, adenosine is quickly metabolized by adenosine deaminase25 that limits its clinical usefulness as a part of combination therapies.
We described previously the synthesis of muramyl dipeptide (D,D) (MDP(D,D)) and nor-muramyl dipeptide (D,D) (nor-MDP(D,D)) derivatives conjugated with adenosine through amino acid spacers as potential immunosuppressants.26 Some of the synthesized compounds exhibited moderate cytotoxic activity against murine leukemia L1210 cells.27 In this study, we report the synthesis of a new series of batracylin derivatives conjugated with adenosine as well as their precursors N-(Nω-amino acid)–BAT with linking synthetic amino acid side chains of different length. In our method, synthesis of new conjugates is based on the reaction of batracylin derivatives with linking synthetic amino acids and 6-chloropurine riboside. We hypothesized that combination of adenosine and batracylin derivatives will result in analogues with improved activity and/or selectivity toward tumor cells. In this paper, we present the method of synthesis of new these analogues and evaluation of their cytotoxic activity as well as biological effects induced by these compounds in tumor cells. Unexpectedly, we have found that at physiological pH in water and media adenosine–amino acid–BAT conjugates form supramolecular structures and that prevents these compounds from entering cells. Consequently, these compounds exert no biological activity. In contrast, several their amino acid–BAT precursors showed greatly enhanced cytotoxic activity compared to BAT and these compounds strongly interfered with DNA topoisomerase II activity and its cellular functions, in particular inhibited centromere segregation during mitosis in drug-treated tumor cells by preventing a topoisomerase II-dependent Aurora B activation.
Results and discussion
Chemistry
We synthesized new analogues of batracylin combined with adenosine as potential anticancer agents. The synthesis of this compounds were carried out according to Schemes 1 and 2. The batracylin (4) was synthesized by our modified method based on the Czerniak–Einhorn reaction. In this method, a symmetrically protected 1,4-phenylenediamine derivative has undergone the Czerniak–Einhorn reaction, and after hydrolysis of the protecting groups, BAT (4) was obtained.28 We synthesized also the batracylin derivatives N-(Nω-Boc-amino acid)–BAT 6a–h, using N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) in anhydrous dichloromethane (DCM).28 We obtained derivatives 6a–h in yields 49–65%. The compounds 6b, 6c, 6e have been published previously.28,29 The compounds 6a, 6d and 6f–h were prepared by the same procedure.28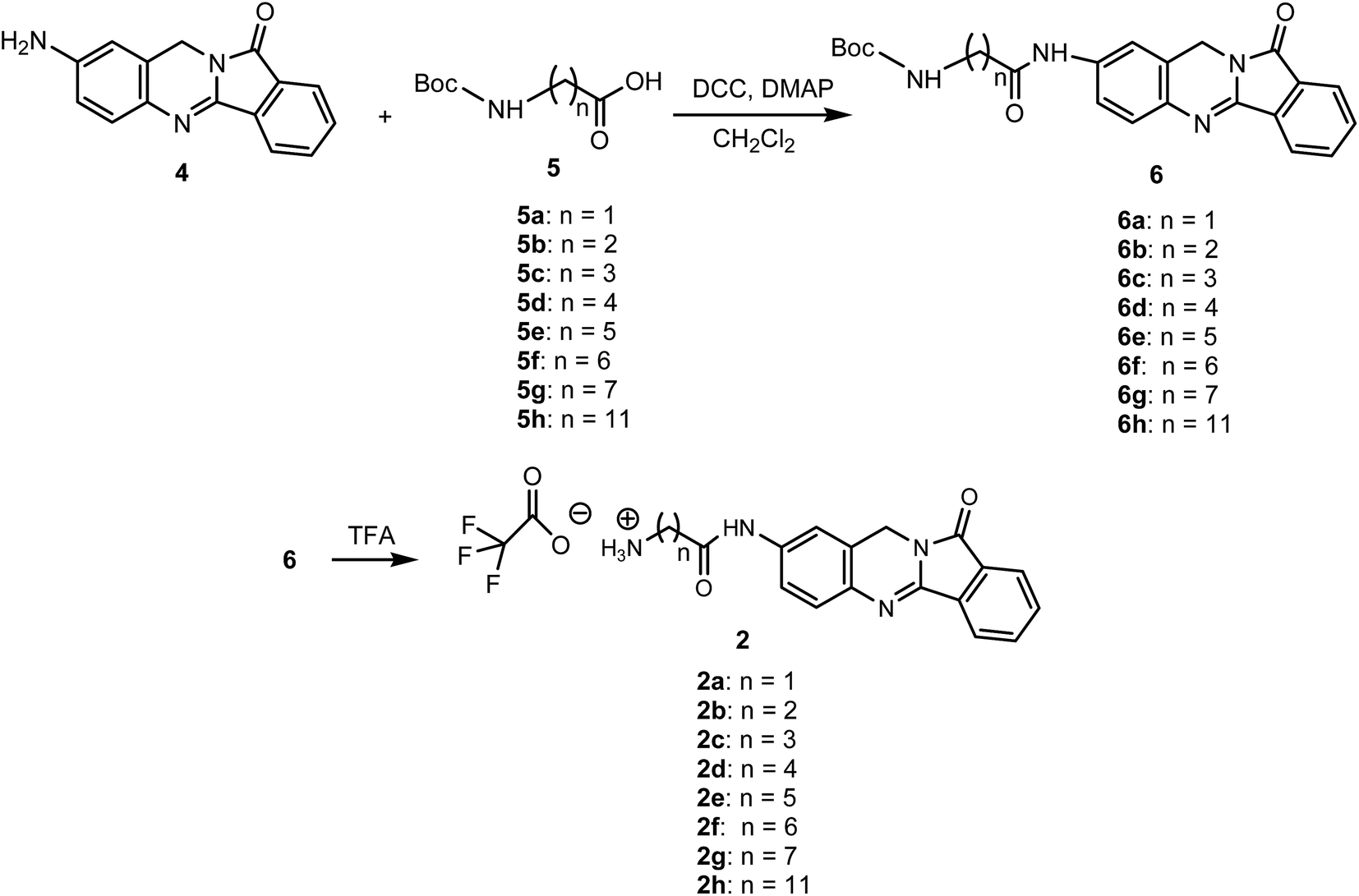 | ||
| Scheme 1 Synthesis of amino acid–BAT derivatives 2a–h. | ||
 | ||
| Scheme 2 Synthesis of adenosine–amino acid–batracylin analogues 3a–h. | ||
The tert-butyloxycarbonyl (Boc) protecting group was removed by treatment with 90% trifluoroacetic acid (TFA) to give the products 2a–h (Scheme 1).30 Reaction of nucleophilic substituted between 6-chloropurine riboside 1 and an amine group of batracylin derivatives 2a–h and N,N-diisopropylethylamine (DIEA) were achieved during reaction in anhydrous ethanol under N2 for 4 h. The reaction was carried out in the boiling solvent. After 4 h EtOH was removed from a reaction by evaporation under vacuum (Scheme 2). The final products 3a–h were purified with preparative TLC, and their identities were confirmed by high resolution 1H NMR (500 MHz), 13C NMR spectroscopy and MALDI-TOF mass spectrometry analysis. The purity of all compounds was evaluated by HPLC and was found to be higher than 95% (see ESI Fig. 1 and 2†).
Our synthesized analogues 2a–h and 3a–h were tested for their cytotoxicity and evaluation of their mechanisms of actions toward tumor cells.
Biological assays
| Compound | IC50 value [μM] | |
|---|---|---|
| A549 cells | HL-60 cells | |
| BAT | 46.3 ± 7.9 | 66.6 ± 4.4 |
| 3a | >100 | >100 |
| 3b | >100 | >100 |
| 3c | >100 | >100 |
| 3d | >100 | >100 |
| 3e | 40.6 ± 3.2 | >100 |
| 3f | >100 | >100 |
| 3g | >100 | >100 |
| 3h | >100 | >100 |
| Riboside | 29.3 ± 1.5 | 48.5 ± 2.5 |
Even more unexpectedly, several of the amino acid–BAT precursors showed a greatly increased cytotoxicity toward both tumor cell types compared to BAT (Table 2). Sensitivity toward both A549 and HL-60 cells was higher from about 2- to more than 25-fold compared to the parent compound. Sensitivity profile for all tested compounds was strikingly comparable in both types of tumor cells e.g. compound 2c was about 17 times more cytotoxic toward A549 cells than BAT and, at the same time, was about 16 times more active toward HL-60 cells than BAT. Similarly, we found that compound 2g was slightly less active than BAT toward both types of tumor cells tested (Table 2).
| Compound | IC50 value [μM] | |
|---|---|---|
| A549 cells | HL-60 cells | |
| BAT | 46.3 ± 7.9 | 66.6 ± 4.4 |
| 2a | 2.7 ± 0.4 | 4.2 ± 0.5 |
| 2b | 35.0 ± 5.9 | 18.6 ± 3.4 |
| 2c | 17.3 ± 3.1 | 15.9 ± 2.7 |
| 2d | 6.3 ± 1.2 | 6.0 ± 0.9 |
| 2e | 7.9 ± 0.5 | 15.9 ± 3.1 |
| 2f | 15.3 ± 1.3 | 26.2 ± 2.4 |
| 2g | 69.7 ± 6.9 | 81.1 ± 5.6 |
| 2h | 1.8 ± 0.2 | 2.4 ± 0.4 |
We hypothesize that observed differences in cytotoxicity of amino acid–BAT conjugates are resulting from the similarity to the natural amino acids side chain structure. Structure of amino acid chain in compound 2a imitate the smallest amino acid – glycine, therefore its mechanism of action relay more on drug–DNA intercalation (ESI Fig. 7†) than drug–protein interactions. Decreased activity of 2b compound can be explain, that there is no natural amino acid with two carbon chain and positively charged group. Both serine and cysteine contain negatively charged groups. Also decreased intercalation to DNA is observed for compound 2b (ESI Fig. 7†) due to larger distortion of flat structure than in 2a derivative. Structure of side chain of lysine can be imitate by the structure of amino acids in compounds from 2c to 2f. Those four compounds have moderate cytotoxic properties and do not strongly intercalate to the DNA, however mimic of the lysine side chain may suggest that derivatives from 2c to 2f interact with enzymes. This hypothesis however, need to be support by further study using molecular modeling methods. Lack of activity of derivative 2g is caused probably by poor lipophilic efficiency (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 2.08 ± 0.90 for 2g and logP 0.69 ± 0.90 for BAT) and for this reason 2g interact forcefully with membrane lipids molecules than proteins or DNA molecules. Compound 2h is a exception because this compounds have very different logD distribution curves than other compounds (ESI Fig. 8†). Derivative 2h is more water than lipid soluble only in acidic pH. Therefore we observed very significant accumulation of this compounds in acidic vesicles (ESI Fig. 1†) and as mentioned later for this reasons we excluded 2h compound from extended studies.
P = 2.08 ± 0.90 for 2g and logP 0.69 ± 0.90 for BAT) and for this reason 2g interact forcefully with membrane lipids molecules than proteins or DNA molecules. Compound 2h is a exception because this compounds have very different logD distribution curves than other compounds (ESI Fig. 8†). Derivative 2h is more water than lipid soluble only in acidic pH. Therefore we observed very significant accumulation of this compounds in acidic vesicles (ESI Fig. 1†) and as mentioned later for this reasons we excluded 2h compound from extended studies.
 | ||
| Fig. 1 Effect of selected batracylin (BAT) and BAT-conjugates on the catalytic activity of purified type I (upper panel) and type II (lower panel) DNA topoisomerases. | ||
HL-60 and A549 cells were treated with BAT and two amino acid–BAT conjugates (compounds 2a and 2d) at doses corresponding to IC90 concentrations and showed significant changes in cell cycle progression (Fig. 2).
 | ||
| Fig. 2 Effect of BAT and its synthetic amino acid conjugates on the cell cycle distribution of tumor cells. Cells were exposed to specified drug doses for the time indicated and analyzed by flow cytometry. * – measured as a fraction of living cells; ** – measured as a fraction of sub-G1 of the total counted events. | ||
After 24 h most of the cells arrested their cell cycle progression in G2/M phases. This block was stable for next 120 h of incubation of A549 cells. However, for HL-60 after long term 120 h exposure a significant increase of cell debris fraction (with hypodiploid DNA content) was observed. When this fraction was re-calculated as a percentage of total events, the fraction changed from less than 1% in control samples to more than 25% for BAT and more than 75% for 2a and 2d derivatives. These results suggested that the prolonged arrest of HL-60 cells in G2/M phases was sufficient to induce apoptosis, that resulted in fragmentation of drug-treated leukemia cells and increased cell debris fraction. Analysis of nuclear morphology of drug-treated HL-60 cells showed that these cells after 24 h of treatment entered mitosis. However, most of these mitoses were abnormal with many aberrant features like multipolar spindle or lagging chromosomes during metaphase to anaphase transition (see ESI Fig. 5†). At later incubation times, a small fraction of drug-treated cells become polyploid (Fig. 2). In contrast to HL-60 cell line, treatment of A549 cells with studied compounds led to G2/M arrest at 24 h followed by increasing fraction of polyploid cells at longer incubation time (120 h) with a marginal fraction of dying cells (Fig. 2 and ESI Fig. 5†).
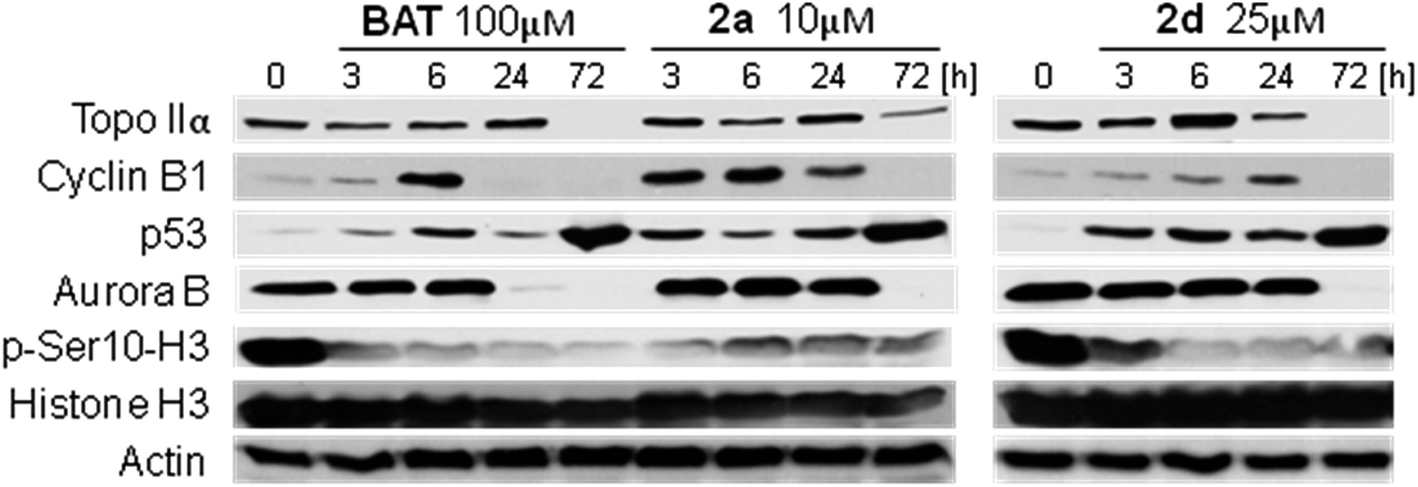 | ||
| Fig. 3 Effect of BAT and its conjugates on the expression of proteins involved in the regulation of cells proliferation and mitotic transit. Cells were exposed to specified drug doses for the time indicated and analyzed by western blotting. | ||
 | ||
| Fig. 4 Induction of double-stranded DNA breaks in A549 cells by BAT and its amino acid conjugates. Cells were exposed to specified drug doses for the time indicated and analyzed by fluorescence microscopy after staining with DAPi (blue) γ-H2AX antibodies (green). Thick arrow shows cell nucleus containing DNA breaks, thin arrow points to cell nucleus of a daughter cell with apparently no DNA breaks. | ||
It was shown previously by Coelho et al. that inhibition of topo II activity by its chemical inhibitor ICRF-187 or silencing topo II expression with siRNA can significantly attenuate Aurora B activity, however this inhibition is only moderate when using ICRF-187 and requires prolonged time (72 h) when using siRNA.36 Strong reduction of histone H3 phosphorylation during whole incubation time, strongly supports the idea that BAT and its amino acid derivatives directly inhibited Aurora B activity or its topo IIα-dependent activation. When the activity of topo IIα and Aurora B is inhibited in cells entering mitosis, mitotic division cannot properly proceed. As it was shown previously, inhibition of Aurora B activity leads to increased number of syntelic as well as mero-amphitelic microtubule attachments.37 A549 cells treated with our compounds arrested during cytokinesis and formed droplet-like structures with lagging DNA between dividing cell nuclei that frequently was associated with an unequal DNA distribution between daughter cells. More importantly, these lagging structures contained centromeric but not telomeric DNA as revealed using FISH analysis with appropriate molecular probes (Fig. 5). Unusual DNA structures were quite stable and were still observed after 72–120 h of drug treatment and followed by accumulation of tumor cells with features of senescent cells, such as enlarged cells wit flat morphology and increased activity of senescence-associated β-galactosidase (see ESI Fig. 6 and 7†). Spindle assembly checkpoint requires Aurora B activity to arrest the cell cycle in mitosis,36 and since activity of Aurora B is inhibited by our compounds, spindle assembly checkpoint cannot be maintained and mitotic division is allowed, even in the presence of DNA damage. Drug-treated tumour cells undergo aberrant cytokinesis at least in part because mid-body complex containing Aurora B protein had abnormal structure (see ESI Fig. 8†). As a result, cytokinesis could not be completed, which explains increased fraction of polyploid cells after 120 h of treatment. Taken together cells treated with our compounds arrest their cell cycle progression in G2 phase and undergo drug induced premature senescence or enter abnormal mitosis followed by cell polyploidization or non-equal DNA distribution between daughter cells associated with the presence of lagging DNA structures. Non-proliferative status of cells treated with studied compounds was evident from western blot analysis since expression of proteins (topo IIα and Aurora B – proliferative markers, cyclin B1 – marker of mitotic exit) involved in cell cycle progression was strongly reduced after 72 h of treatment (Fig. 3). Loss of Aurora B kinase activity by batracylin and its amino acid conjugates may be due to a direct inhibition on as another author says by indirect interactions with topoisomerase IIα.
 | ||
| Fig. 5 Nuclear morphology of cells treated with BAT and its amino acid conjugates. Cells were exposed for 3h to studied compounds and analysed with FISH as described in Materials and methods followed by fluorescence confocal microscopy. Blue: DAPI staining of DNA; green – telomeric DNA probe and red – centromeric DNA probe. | ||
Together, our study led to the synthesis of original group of BAT analogues 2a–h and 3a–h with potential increased antitumor activity and improved pharmacological properties, including potentially lower general toxicity. After evaluation in further pre-clinical studies in vivo can be proposed as new antitumor agents. Unfortunately, functionalization of BAT by addition of adenosine with a synthetic amino acid linker led to conjugates which showed no biological activity due to formation of supramolecular structures. However, BAT–amino acid derivatives were up to 25-fold more cytotoxic toward tumor cells compared to the parent BAT. Our result showed that BAT and its new BAT–amino acid derivatives produced DNA damage and strongly interfered with centromere separation in tumor cells as a result of the simultaneous inhibition of DNA topoisomerase IIα and Aurora B activation.
Conclusions
A series of non-protein amino acid–batracylin derivatives 2a–h (Scheme 1) and its conjugates with adenosine 3a–h (Scheme 2) were synthesized as potential anticancer compounds. These compounds were tested for their cytotoxicity and evaluation of their mechanisms of actions toward two human cell lines lung adenocarcinoma (A549) and human leukemia (HL-60).Several of the amino acid–BAT derivatives showed greatly increased cytotoxicity against both tumor cell types compared to BAT. Sensitivity toward both A549 and HL-60 cells was higher from about 2- to more than 25-fold compared to the BAT e.g. compound containing 12-aminododecanoic acid was about 30 times more cytotoxic against A549 and HL-60 cells than BAT. These compounds strongly interfered with DNA topoisomerase II activity and its cellular functions. In particular, these conjugates inhibited centromere segregation during mitosis in drug-treated tumor cells by preventing a topoisomerase II-dependent Aurora B activation.
The majority of BAT–amino acid–adenosine conjugates were biologically inactive toward both A549 as well as HL-60 cells. Only compound containing 6-aminohexanoyl linker was specifically cytotoxic against A549 cells but indicated comparable activity to BAT. Unexpectedly, we have found that in water and culture media adenosine–amino acid–BAT conjugates form supramolecular structures and that prevents these compounds from entering cells. The molecular mechanism responsible for this unexpected formation of supramolecular structures in water solutions is not clear at the moment and requires further studies.
Several received under the paper compounds characterized by promising biological properties, requiring further research and have potential application.
Experimental
Materials and methods
Melting points (mp uncorrected) were determined on the Kofler-block apparatus. All chemicals and solvents were of reagent grade and were used without further purification. The reactions were monitored by TLC on Merck F254 silica gel pre-coated plates. The following solvent systems (by vol.) were used for TLC development: CH2Cl2–MeOH (7:1, v/v) (A), CH2Cl2–MeOH (10:1, v/v) (B), CH2Cl2–MeOH (20:1, v/v) (C). CH2Cl2–MeOH (50:1, v/v) (D), CH2Cl2–(CH3)2CO (1:1, v/v) (E), CH2Cl2–MeOH–TEA (40:1:0.1 v/v) (F).
MS spectra were recorded on matrix-assisted laser desorption/ionization-time on flight mass spectrometry (MALDI-TOF MS, Biflex III Bruker) (samples: 3a, 3b, 3d, 3f–h) and with a 6540 UHD Accurate-Mass Q-TOF spectrometer using the negative ESI mode (samples: 3c, 3e, 6a, 6d, 6f–h).
Proton and carbon NMR spectra were recorded in dimethyl sulfoxide-d6 (DMSO-d6) (isotopic enrichment 99.95%) solutions at 293 K and 300 K (sample 3h) using a Varian Unity 500 Plus spectrometer (500.13 MHz for 1H, 125.76 MHz for 13C) with a sample concentrations collected in Table 1, using 5 mm inverse detection broadband probes and deuterium lock. The central peak of DMSO-d6 signals (2.49 ppm for 1H and 39.50 ppm for 13C) was used as the internal reference standard. Two-dimensional 1H spectra were measured in the phase-sensitive mode with a spectral width of 8000 Hz and 30272 data points providing a digital resolution of 0.529 Hz per point. The 13C spectra were measured with the spectral width of 28777 Hz and 74816 data points providing a digital resolution of 0.769 Hz per point, relaxation delay 1.5 s. The structures of compounds 3d and 3h were confirmed additionally by 2D g-COSY and ROESY 1H NMR (see ESI Table 1†). The g-COSY spectrum was acquired in 4096 × 320 matrix with eight accumulations per increment and was processed in a 4K × 1K matrix. The ROESY spectrum was acquired with a mix time of 350 ms in a 2560 × 300 matrix with 8 accumulations per increment in a 4K × 1K matrix. HSQC and HMBC experiments were performer with pulse field gradients. The HSQC spectrum was acquired in the phase-sensitive mode. The spectral windows for 1H and 13C axes were 5.169 Hz and 18.853 Hz, respectively. The data were collected in a 2048 × 187 matrix and processed in a 2K × 1K matrix. The HMBC spectrum was acquired in absolute value mode. The spectral windows for 1H and 13C axes were 5.169 Hz and 21.995 Hz, respectively. The data were collected in a 2560 × 210 matrix and processed in a 2K × 1K matrix.
The purity of all compounds was evaluated by HPLC using Agilent liquid chromatograph series 1290 (Agilent Technology, Waldbronn, Germany) consisting of binary pump G4220A, autosampler G4226A, thermostated column compartment G1316C, diode-array detector G1315C. Chromatographic column: Supelcosil C-18, (4.6 × 150 mm), 3 μm, Supelco. Analytes were dissolved in a mixture of DMSO/MeOH (1:1; v/v) and 2 μL of the solution were injected onto the chromatographic column. A mixture of A: 0.1% HCOOH in water B: 0.1% HCOOH in AcCN/MeOH (1:1; v/v) was used as a mobile phase in gradient mode at flow rate of 2.0 mL min−1. Gradient: at 5 min 20% B; at 15 min 100% B for analyzed compounds. The UV-vis detector was operated at 254, 210 and 580 nm (DAD in single wavelength mode). All analyses were done at 40 °C.
8-Aminoisoindolo[1,2-b]quinazolin-12(10H)-one (BAT) 4
This was prepared according to the method described from N-[2-(phthalimidomethyl)-1,4-acetyl]phenylenediamine (8) and recrystallized from N,N-dimethylformamide (DMF).28Batracylin derivatives [N-(Nω-Boc-amino acid)–BAT] 6a–h
The procedure for the synthesis of compounds 6b, 6c, 6e have been published previously.28 The compounds 6a, 6d and 6f–h were prepared by the same procedure. The Boc-protecting groups were removed by treatment with TFA and then coupling gave compounds 2a–h.General procedure for synthesis of adenosine analogues (3a–h)
Under a nitrogen atmosphere, a mixture of 6-chloropurine riboside (1) (1 mmol), batracylin derivatives (2a–h) (1 mmol) and DIPEA (10 mmol) in anhydrous EtOH (4 mL) was stirred at boiling point of solvent for 4 h. The reaction was monitored with TLC in system solvent A. After evaporating the solvent in vacuo, product was purified by TLC in solvent systems B and C.Cell culture
Cell lines were purchased from American Type Culture Collection (Rockville, MD). The cells A549 and HL-60 were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics (100 units per mL penicillin and 100 μg mL−1 streptomycin) and cells were cultivated at 37 °C in 5% CO2/95% air atmosphere. Cell lines were screened routinely for mycoplasma by the PCR method with Mycoplasma Plus PCR primer set (Stratagene, La Jolla, CA).Cytotoxicity assay
The cytotoxicity was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, exponentially growing cells were continuously exposed to different drug concentrations and the cellular viability was evaluated after 120 h. Cells were exposed to the MTT tetrazolium salt for 4 h at 37 °C, and the formation of formazan was measured after solubilization in 1 mL DMSO by a ASYS UVM340 microplate reader (Biochrom Ltd.). The concentrations required to inhibit cell growth by 50% compared to untreated controls were determined from the curves plotting survival as a function of dose by use of the GraphPad Prism 5 program. All values are averages of at least three independent experiments, each done in duplicate.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) D and logP of studied compounds.
The protonation at different pH was calculated for BAT and its derivatives using the SPARC online calculator (ARChem – Automated Reasoning in Chemistry) at http://archemcalc.com/sparc/. The logD curves were created using ChemAxon online calculator (at https://www.chemaxon.com). The logP values were calculated using ACD/logP Freeware (component of ACD/ChemSketch Freeware program).
D and logP of studied compounds.
The protonation at different pH was calculated for BAT and its derivatives using the SPARC online calculator (ARChem – Automated Reasoning in Chemistry) at http://archemcalc.com/sparc/. The logD curves were created using ChemAxon online calculator (at https://www.chemaxon.com). The logP values were calculated using ACD/logP Freeware (component of ACD/ChemSketch Freeware program).
Flow cytometry analysis of cell cycle distribution
Distribution in different phases of the cell cycle was analyzed by flow cytometry after treatment of tumor cells with studied drugs. Briefly, following drug treatment with a dose corresponding to IC90, cells were washed in ice-cold phosphate buffered saline (PBS) and fixed in 70% ethanol at −20 °C. Cells were stained in PBS containing 20 μg mL−1 propidium iodide and 100 μg mL−1 ribonuclease A for 30 min at room temperature. Samples were analyzed by a Guava EasyCyte™ 8 flow cytometer (Merck-Millipore) equipped with a 488 nm laser, and the distribution of cells in the cell cycle was calculated using MultiCycle software (Phoenix Flow Systems).Inhibition of DNA relaxation mediated by DNA topoisomerases
Supercoiled DNA plasmid pBR322 (>95% form I) was purchased from Thermo Scientific, human type I topoisomerase was purchased from MoBiTec and human type IIα topoisomerase was purchased from TopoGen. The reaction mixture contained 200 ng of pBR322 DNA in reaction buffer (20 mM Tris–HCl, pH 7.5, 7.5 mM MgCl2, 0.5 mM DTT, 150 mM KCl, 1 mM ATP) as well as studied compounds. The reaction was initiated by the addition of topoisomerase IIα enzyme and allowed to proceed at 37 °C for 30 min. Reactions were terminated by addition of a loading buffer (0.1% SDS, 0.05% bromophenol blue, 2.5 mM EDTA, 10% sucrose, final concentrations). The samples were separated in 1% agarose gels at 0.5 V cm−1 for 18 h in TBE buffer (90 mM Tris-base, 70 mM boric acid, 1 mM EDTA, pH 8). Gels were stained with 0.5 μg mL−1 ethidium bromide to visualize DNA and photographed under UV illumination. The conditions for DNA relaxation assay for type I topoisomerase were the same except different reaction buffer was used (10 mM Tris–HCl, pH 7.5, 7.5 mM MgCl2, 60 mM KCl).Nuclear and cellular morphology of tumor cells
Cells were attached to cover slides in 35 mm Petri dishes and treated with studied compounds for different time periods. Following drug treatment, cells were stained with 1 μg mL−1 Hoechst 33342 alone or with 1 μg mL−1 Hoechst 33342 and 7.5 μg mL−1 fluorescein diacetate dyes for 15 min and analyzed by Olympus BX-60 fluorescent microscope equipped with respective optical filters. Images were recorded using XC50 digital camera and image acquisition software CellSens.Immunofluorescence
Cells were fixed with ice cold methanol, blocked with 5% BSA/0.05% Tween-20/PBS for 1 h at 37 °C, and incubated with anti-Aurora B diluted 1:000 in 5% BSA/PBS (Abcam, ab2254) overnight at 4 °C. An anti-rabbit Alexa Fluor 488 (BioLegend, 406416) was used as the secondary antibody. For detection of H2AX phosphorylation, cells were fixed with 3.7% PFA, permeabilized with 0.2% Triton X-100 and blocked with 5% BSA/PBS for 10 min per RT. Slides were incubated overnight at 4 °C with anti-phospho-(S139)-H2AX Alexa Fluor 488 conjugated antibody diluted 1:200 in 5% BSA/PBS (BioLegend, 613406). DAPI was used to stain the cell nuclei at a concentration 0.5 μg mL−1. Images were analyzed using LSM 700 confocal microscope (Zeiss).
Fluorescence in situ hybridization (FISH) assay
The FISH assay was performed according to manufacturer instructions. Cells where fixed in methanol:acetic acid solution 3:1 and dehydrated through a series of ethanol solutions (70, 90 and 100%) and air-dried. Slides were then baked at 65 °C and after cooling, washed with acetone for 10 min and air-dried. RNA was digested by incubation of samples with RNase A (100 μg mL−1 in 2× SSC) at 37 °C for 1 h. After 2 washes with PBS and 2× SSC buffer, slides were incubated with pepsin (0.5 μg mL−1 in 10 mM HCl) for 3 min at RT, then washed with PBS and dehydrated in ethanol as described above. DNA was denatured by incubation of slides in 70% formamide/2× SSC solution at 70 °C for 2 min and instantly cooled to −20 °C by immersion in a cold 70% ethanol/water solution. FITC-conjugated pan-telomeric chromosome (starFISH, 1696-F-02) and biotin-conjugated pan-centromeric chromosome (starFISH, 1695-B-02) probes were used (Cambio). Probes for centromeric and telomeric DNA were diluted in a buffer provided by the manufacturer, denatured at 85 °C for 10 min and rapidly cooled on ice. Hybridization under coverslips was carried out at 37 °C overnight in a humidified chamber. Cover glasses were then removed by immersion in 2× SSC at 37 °C and non-hybridized probes were washed off by incubation in 50% formamide/2× SSC at 37 °C. Blocking step with 5% BSA/0.05% Tween-20/PBS was performed and then slides were incubated with rabbit anti-FITC antibodies (1:200, Invitrogen, 71-1900) and mouse-anti-biotin Alexa Fluor 594-conjugated antibodies (1:100, Jackson ImmunoResearch Labs, 200-582-211) for 1 h at 37 °C. Samples were washed three times in 0.05% Tween 20/PBS and incubated 1 h at 37 °C with donkey anti-rabbit Alexa Fluor 488-conjugated antibodies (1:100, Thermo Fisher Scientific, SA5-10038). DAPI was used to stain the cell nuclei at a concentration 0.5 μg mL−1 in water. Images were analyzed by Leica confocal microscope TCS SP8 X equipped with the white light laser and appropriate filters (Leica Microsystems).
SA-β-galactosidase staining
Cells were treated with studied compounds with the IC90 doses for 120 h and washed in PBS, fixed for 3–5 min (room temperature) in 3.7% formaldehyde, washed, and incubated at 37 °C (no CO2) with freshly prepared staining solution: 1 mg mL−1 of 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal) in 40 mM citric acid/sodium phosphate, pH 6.0, (containing 5 mM potassium ferrocyanide; 5 mM potassium ferricyanide; 150 mM NaCl; 2 mM MgCl2) for 12–16 h. Images were analyzed by Olympus BX-60 microscope and recorded using XC50 digital camera as well as image acquisition software CellSens.Western blot analysis
Cells were harvested on ice by scrubbing with RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was determined using BCA method (Price) and 50 μg of total protein was loaded onto SDS-PAGE gel and blotted on the PVDF membrane. Following blocking step in 5% BSA/TBST membranes where incubated with primary antibodies toward topoisomerase IIα (BioTrend), cyclin B1 (Santa Cruz), p53 (Santa Cruz), Aurora B (Abcam), p-Ser10-H3 (Sigma), histone H3 (Cell Signaling), actin (Santa Cruz) overnight at 4 °C. Membranes were washed tree times with TBST and incubated with indicated secondary antibody anti-mouse-HRP, anti-rabbit-HRP, anti-goat-HRP (Jacson ImmunoResearch) for 1 h at RT. Membranes where visualized by ECL (Thermo) and developed using X-ray films and developing machine (Agfa).Acknowledgements
This work was financially supported by the National Science Center (Poland), grant no. 2012/05/B/NZ7/02461. The authors would like to thank Dr Michał Rychłowski (Laboratory of Viral Molecular Biology, Intercollegiate Faculty of Biotechnology, Gdansk) for help with confocal microscope analyses.References
- R. V. Chari, Acc. Chem. Res., 2008, 41, 98 CrossRef CAS PubMed.
- J. Plowman, K. D. Paull, G. Atassi, S. D. Harrison, D. J. Dykes, H. J. Kabbe, V. L. Narayanan and O. C. Yoder, Invest. New Drugs, 1988, 6, 147 CrossRef CAS PubMed.
- W. R. Waud, S. D. Harrison, K. S. Gilbert, W. R. Laster and D. P. Griswold, Cancer Chemother. Pharmacol., 1991, 27, 456 CrossRef CAS PubMed.
- P. Mucci-LoRusso, L. Polin, M. C. Bissery, F. Valeriote, J. Plowman, G. D. Luk and T. H. Corbett, Invest. New Drugs, 1989, 7, 295 CrossRef CAS PubMed.
- G. Atassi, P. Dumont, H. J. Kabbe and O. Yoder, Drugs Exp. Clin. Res., 1988, 14, 571 CAS.
- Y. Luo, Y. F. Ren, T. C. Chou, A. Y. Chen, C. Yu, L. F. Liu and C. C. Cheng, Pharm. Res., 1993, 10, 918 CrossRef CAS.
- V. A. Rao, K. Agama, S. Holbeck and Y. Pommier, Cancer Res., 2007, 67, 9971 CrossRef CAS PubMed.
- M. M. Ames, D. A. Mathiesen and J. M. Reid, Invest. New Drugs, 1991, 9, 219 CrossRef CAS PubMed.
- G. J. Stevens, J. L. Burkey and C. A. McQueen, Cell Biol. Toxicol., 2000, 16, 31 CrossRef CAS PubMed.
- L. W. Wormhoudt, J. N. M. Commandeur and N. P. E. Vermeulen, Crit. Rev. Toxicol., 1999, 29, 59 CrossRef CAS PubMed.
- S. Kummar, M. E. Gutierrez, L. W. Andrson, R. W. Klecker, A. Chen, A. J. Murgo, J. H. Doroshow and J. M. Collins, Cancer Chemother. Pharmacol., 2013, 72, 917 CrossRef CAS PubMed.
- K. Dzierzbicka, W. Januchta and A. Skladanowski, Curr. Med. Chem., 2012, 19, 4475 CrossRef CAS PubMed.
- J. Roservear and F. K. Wilshire, Aust. J. Chem., 1990, 43, 339 CrossRef.
- V. Stella, L. A. Al-Razzak and H. J. Kabbe, U.S. Pat., 4.980.343, 1990.
- H. G. Larchen, N. Piel, J. P. Lorentzen, H. P. Antonicek and H. J. Kabbe, European Patent Office No. 0 501 250 A1, 1992Chem. Abstr., 1993, 118, 60133k.
- V. Stella, L. A. Al-Razzak and H. J. Kabbe, C.A. Patent 2.001.494, 1990.
- J. Chen, H. Neumann, M. Beller and X. Wu, Org. Biomol. Chem., 2014, 12, 5835 CAS.
- M. C. Tseng, P. Y. Lai, L. Shi, H. Y. Li, M. J. Tseng and Y. C. Chu, Tetrahedron, 2014, 70, 2629 CrossRef CAS.
- V. Ralevic and G. Burnstock, Pharmacol. Rev., 1998, 50, 413 CAS.
- B. B. Fredholm, A. P. Jzerman, K. A. Jacobson, K. N. Klotz and J. Linden, Pharmacol. Rev., 2001, 53, 527 CAS.
- S. Gessi, K. Varani, S. Merighi, E. Fogli, V. Sacchetto, A. Bennini, E. Leung, S. Mac-Lennen and P. A. Borea, Purinergic Signalling, 2007, 3, 109 CrossRef CAS PubMed.
- M. Samsel and K. Dzierzbicka, Pharmacol. Rep., 2011, 63, 601 CrossRef CAS PubMed.
- J. T. Merrill, C. Shen and D. Schreibmab, Arthritis Rheum., 1997, 40, 1308 CAS.
- H. Kamiya, T. Kanno, Y. Fujita, A. Gotoh, T. Nakano and T. Nishizaki, Cell. Physiol. Biochem., 2012, 29, 687 CrossRef CAS PubMed.
- P. E. Zarek, C. T. Huang, E. R. Lutz, J. Kowalski, M. R. Horton, J. Linden, C. G. Drake and J. D. Powell, Blood, 2008, 1, 251 CrossRef PubMed.
- K. Dzierzbicka, A. Wardowska and P. Trzonkowski, Curr. Med. Chem., 2011, 18, 2438 CrossRef CAS PubMed.
- M. Samsel, K. Dzierzbicka and P. Trzonkowski, Bioorg. Med. Chem. Lett., 2014, 24, 3587 CrossRef CAS PubMed.
- K. Dzierzbicka, P. Trzonkowski, P. Sewerynek and A. Myśliwski, J. Med. Chem., 2003, 46, 978 CrossRef CAS PubMed.
- P. Trzonkowski, K. Dzierzbicka, J. Bociewicz, E. Szmit and A. M. Myśliwski, Int. Immunopharmacol., 2005, 5, 241 CrossRef CAS PubMed.
- K. Dzierzbicka, P. Sowiński and A. M. Kołodziejczyk, J. Pept. Sci., 2006, 12, 670 CrossRef CAS PubMed.
- W. Januchta, M. Serocki, K. Dzierzbicka, G. Cholewiński and A. Skladanowski, Eur. J. Med. Chem., 2015, 106, 85 CrossRef CAS PubMed.
- M. Hofer, L. Dušek, Z. Hoferová, L. Stixová and M. Pospíšil, Physiol. Res., 2011, 60, 913 CAS.
- T. Eckle, E. M. Kewley, K. S. Brodsky, E. Tak, S. Bonney, M. Gobel, D. Anderson, L. E. Glover, A. K. Riegel, S. P. Colgan and H. K. Eltzschig, J. Immunol., 2014, 192, 1249 CrossRef CAS PubMed.
- H. Wang, Y. Mao, N. Zhou, T. S. Hsieh and L. F. Liu, J. Biol. Chem., 2001, 276, 15990 CrossRef CAS PubMed.
- A. Celeste, O. Fernandez-Capetillo, M. J. Kruhlak, D. R. Pilch, D. W. Staudt, A. Lee, R. F. Bonner, W. M. Bonnier and A. Nussenzweig, Nat. Cell Biol., 2003, 5, 675 CrossRef CAS PubMed.
- P. A. Coelho, J. Queiroz-Machado, A. M. Carmo, S. Moutinho-Pereira, H. Maiato and C. E. Sunkel, PLoS Biol., 2008, 26, e207 Search PubMed.
- S. Hauf, R. W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. vanMeel, C. L. Rieder and J. M. Peters, J. Cell Biol., 2003, 161, 281 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Analytical and spectroscopic data for synthesized compounds as well as additional figures. See DOI: 10.1039/c6ra04957e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |