
KMnO4/AcOH-mediated C3-selective direct arylation of coumarins with arylboronic acids†
Jin-Wei Yuan*,
Liang-Ru Yang,
Qiu-Yue Yin,
Pu Mao and
Ling-Bo Qu*
School of Chemistry & Chemical Engineering, Henan University of Technology, Zhengzhou 450001, P.R. China. E-mail: yuanjinweigs@126.com; Fax: +86-371-67756718; Tel: +86-371-67756718
First published on 5th April 2016
Abstract
An efficient protocol for KMnO4/AcOH-mediated dehydrogenative direct radical arylation of coumarins with arylboronic acids to afford 3-arylcoumarin derivatives is described. A similar reaction system is also applicable to the 3-arylation of quinolinone derivatives. These KMnO4/AcOH-mediated coupling reactions occur regioselectively at the C3 position of coumarins and quinolinones. Some notable features of this method are high efficiency, moderate to good yield, and a broad group tolerance.
Introduction
Coumarins are significant natural products, which display wide and interesting pharmacological properties such as anti-breast cancer, anti-HIV, anti-Alzheimer, vasorelaxant and anti-aggregatory activities.1 Coumarin derivatives have proven to be useful skeletons in organic synthesis as valuable building blocks and in pharmaceuticals due to their biological and physical properties. In particular, 3-arylcoumarin represents an important structural element in a selective monoamine oxidase B (MAO-B) inhibitor,2 a horseradish peroxidase (HRP) inhibitor,3 an inhibitor of HIV-1 replication4 (Scheme 1). As a result of their expressive structural diversity, 3-arylcoumarins have been found some possible applications in treatment of allergic disorders, as HSP90 C-terminal inhibitors and strong inhibitors of antigen-induced RBL-2H3 mast cell degranulation.5 Moreover, 3-arylcoumarin derivatives are also important class of fluorophores and found their application as fluorescent labels in complex biological systems.6 Despite the importance of 3-arylcoumarins, the synthesis of 3-arylcoumarins is difficult due to the regioselective bias of 4-arylation of coumarins.7 Thus, the development of novel, efficient, and regioselective methods for the construction of 3-arylcoumarins will be of great value in screening of novel functional active molecules.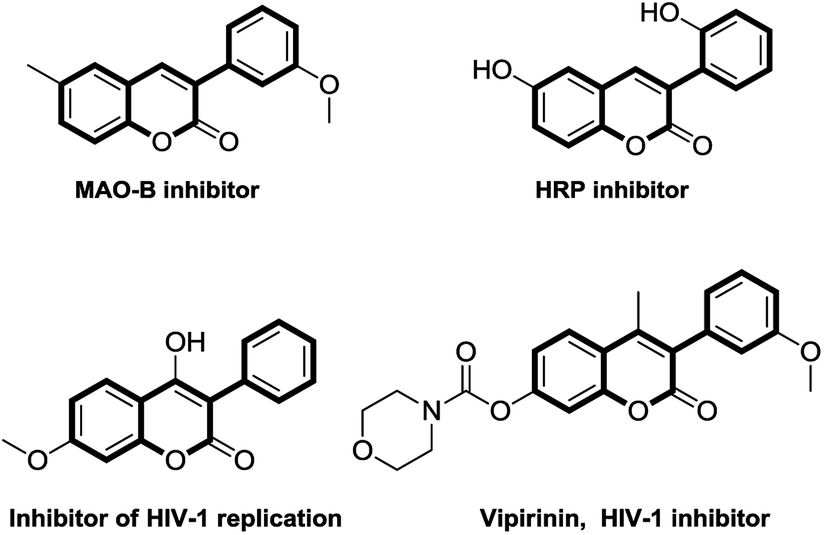 | ||
| Scheme 1 Examples of 3-arylcoumarins medical intermediate. | ||
The general methods for the preparation of 3-arylcoumarins were based on the palladium-catalyzed Suzuki-type coupling reaction or Heck reaction. Matos group reported a method toward 3-arylcoumarins by the Suzuki coupling reaction of 3-chlorocoumarin and phenylboronic acid in the presence of palladium complex and sodium carbonate at 110 °C for 2–3 h in 55–65% yields.8 Wu group synthesized 3-arylcoumarins using 3-bromocoumarin derivatives and phenylboronic acid in the presence of palladium complex and base, and Matsuura group used 3-bromocoumarin and aromatic compounds by photo-catalyzed coupling reaction to obtain 3-arylcoumarin derivatives.9 Jafarpour and Messaoudi groups described a synthetic method for the preparation of 3-arylcoumarin by a palladium-catalyzed decarboxylative coupling of coumarin-3-carboxylic acids and aryl halides.10 Knochel group used the 3-zincated coumarin and 4-substituted iodobenzenes through Pd-catalyzed Negishi cross-coupling reaction to obtain 3-arylcoumarins (Scheme 2a).11 Direct functionalization through metal-catalyzed double C–H activation reactions have began to emerge as an alternative route for C–C bond formation.12 A direct arylation approach allows for the construction of C–C bonds without the need for prior functionalization of coupling partners via metalation. A synthetic method of 3-arylcoumarins was described using coumarins and arenesulfonyl chlorides or sodium arenesulfinates via palladium-catalyzed direct C–H functionalization, Cu(OAc)2 as the oxidant for 24 h.13 Yadav group described a direct 3-arylation of coumarins by the reaction of coumarins and phenylhydrazine using K2CO3 as the base in DMSO solvent for 4–24 h.14 You group reported synthesis of 3-arylcoumarins to use coumarins and aromatic compounds by Pd(OAc)2-catalyzed coupling reaction and (NH4)2S2O8 for the oxidant in TFA for 24 h.15 Jafarpour group described a Pd(OAc)2-catalyzed dehydrogenative 3-arylaction of coumarins using coumarins and aryl compounds for the materials, and trifluoroacetic acid anhydride (TFAA) as the solvent at 120 °C for 16 h.16 3-Arylcoumarins were also achieved by Pd(PPh3)4-catalyzed Heck coupling reactions between coumarins and aryliodides using AgOAc for the base for 72 h (Scheme 2b).17 Other procedures for the synthesis of 3-arylcoumarins involving cyclization reactions such as Pechmann, or Perkin reactions have been published as well.18 Although all the above methodologies have been utilized effectively for 3-arylcoumarins, some problems exist with these procedures, such as: (1) the prefunctionalization, and narrow scope of substrates; (2) use of toxic ligands, strong acid solvent, high temperatures, long reaction time and poor yields. Therefore, developing a general and applicable strategy for a variety of 3-arylcoumarins is highly desirable.
 | ||
| Scheme 2 Synthesis of 3-arylcoumarins and 3-arylquinolines. | ||
Recently, radical C–H functionalization of innately reactive heterocycles has re-emerged as an avenue for selective, early or late stage functionalization of pharmaceutically important precursors and products.19 Metal-promoted radical reactions have been achieved substantially utilizing a variety of aryl coupling partners, in which one of the well-known examples of this application is the Mn(OAc)3-mediated reaction. Manganese(III) acetate is a one-electron oxidant, largely used as a radical generator that can lead to C–C bond forming reactions.20 But, manganese(III) acetate is not stable, and it occurs easily disproportionated reaction to manganese(II) and manganese(IV). The potassium permanganate/acetic acid system in an organic solvent is a powerful substitute for manganese(III) acetate.21 Guided by recent studies on manganese(III)-promoted hemolytic aromatic substitution (HAS) using arylboronic acid as C-radical precursors, we decided to investigate the modular synthesis of 3-arylcoumarins by this approach. Herein, we disclose a KMnO4/AcOH-mediated dehydrogenative direct and regioselective radical arylation of coumarins with arylboronic acids to afford 3-arylcoumarin derivatives in good to high yield (Scheme 2c).
Results and discussion
Our initial experiments showed that using a PdCl2 or Pd(OAc)2 as the catalyst without the oxidant, coumarin 1a occurred with phenylboronic acid 2a to obtain 3-phenyl coumarin 3a with a low yield (43% and 22%) (Table 1, entries 1 and 2). Various oxidants including CuSO4, FeCl3·6H2O, KMnO4, MnO2, MnSO4, and Mn(OAc)2 were added to the reaction system without the catalyst, respectively. CuSO4, MnSO4, and Mn(OAc)2 were found to be ineffective (Table 1, entries 3, 7 and 8). When FeCl3·6H2O, KMnO4, and MnO2 were used however selective phenylation of coumarin at C-3 was pleasingly achieved albeit in 32%, 40%, and 28% (Table 1, entries 4–6). Among various oxidants screened, KMnO4 proved to the most effective. This fact showed that the oxidant was crucial to the coupling efficiency in C–H activation reactions. Subsequently, only the oxidant KMnO4 was used to promote the reaction without any catalyst. The solvent also affected the coupling reaction of coumarin and phenylboronic acid. No product was found with C2H5OH, DCE, DMF or dioxane as solvent (Table 1, entries 9–12), and only low yield was obtained when CH3CN was employed (Table 1, entry 5), where AcOH turned out to be the most appropriate (Table 1, entry 13). The amount of KMnO4 was also screened. To our delight, the yield was improved to 75% when 2.0 eq. KMnO4 was used, which indicated that the KMnO4 played an important role in this reaction (Table S1, ESI†). The ratio of substrates coumarin and phenylboronic acid was investigated, and the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 of coumarin and phenylboronic acid proved to be best result (Table S2, ESI†). When the reaction temperature was increased from 20 °C to 80 °C, the yield of 3a was enhanced from 30% to 80% (Table 1, entries 14–18). However, the product yields dramatically dropped if the reaction temperature continued to be increased, and 80 °C was found to be best choice. Various reaction times were also examined, 0.5 h proved to the best appropriate and the yield was 85% (Table 1, entries 18–21).
:2 of coumarin and phenylboronic acid proved to be best result (Table S2, ESI†). When the reaction temperature was increased from 20 °C to 80 °C, the yield of 3a was enhanced from 30% to 80% (Table 1, entries 14–18). However, the product yields dramatically dropped if the reaction temperature continued to be increased, and 80 °C was found to be best choice. Various reaction times were also examined, 0.5 h proved to the best appropriate and the yield was 85% (Table 1, entries 18–21).
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant (eq.) | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: coumarins 1a (0.5 mmol, 73 mg), phenylboronic acid 2a (1.0 mmol, 122 mg), oxidant in solvent (20 mL).b Isolated yield.c PdCl2 (0.1 eq.) was used as the catalyst.d Pd(OAc)2 (0.1 eq.) was used as the catalyst. | |||||
| 1c | — | CH3CN | 90 | 1.0 | 43 |
| 2d | — | CH3CN | 90 | 1.0 | 22 |
| 3 | CuSO4 (0.2) | CH3CN | 90 | 1.0 | 0 |
| 4 | FeCl3·6H2O (0.2) | CH3CN | 90 | 1.0 | 32 |
| 5 | KMnO4 (0.2) | CH3CN | 90 | 1.0 | 40 |
| 6 | MnO2 (0.2) | CH3CN | 90 | 1.0 | 28 |
| 7 | MnSO4 (0.2) | CH3CN | 90 | 1.0 | 0 |
| 8 | Mn(OAc)2 (0.2) | CH3CN | 90 | 1.0 | 0 |
| 9 | KMnO4 (0.2) | C2H5OH | 80 | 1.0 | 0 |
| 10 | KMnO4 (0.2) | DCE | 90 | 1.0 | 0 |
| 11 | KMnO4 (0.2) | DMF | 120 | 1.0 | 0 |
| 12 | KMnO4 (0.2) | Dioxane | 90 | 1.0 | 0 |
| 13 | KMnO4 (0.2) | AcOH | 120 | 1.0 | 50 |
| 14 | KMnO4 (2.0) | AcOH | 20 | 1.0 | 30 |
| 15 | KMnO4 (2.0) | AcOH | 40 | 1.0 | 55 |
| 16 | KMnO4 (2.0) | AcOH | 60 | 1.0 | 62 |
| 17 | KMnO4 (2.0) | AcOH | 80 | 1.0 | 80 |
| 18 | KMnO4 (2.0) | AcOH | 100 | 1.0 | 78 |
| 19 | KMnO4 (2.0) | AcOH | 80 | 0.25 | 70 |
| 20 | KMnO4 (2.0) | AcOH | 80 | 0.5 | 85 |
| 21 | KMnO4 (2.0) | AcOH | 80 | 2.0 | 78 |

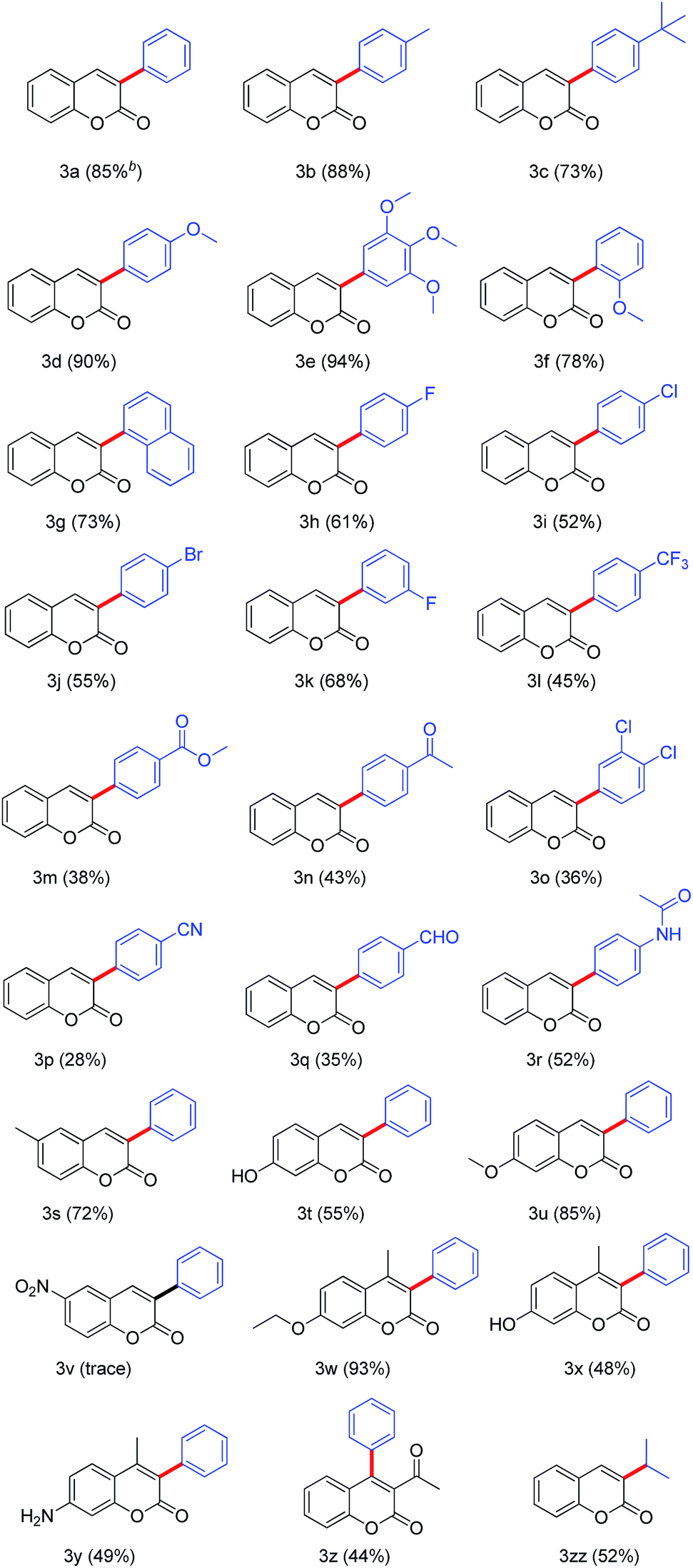
With the optimum conditions in hand (Table 1, entry 20), we next sought to explore the scope of coumarins and arylboronic acids reaction for the construction of 3-arylcoumarins (Table 2). Accordingly, coumarin and various substituted arylboronic acids possessing electron-donating and withdrawing groups were employed in the reaction (Table 2, 3a–r). The results showed that arylboronic acids with various groups including alkyl, methoxy, halogeno, carbonyl, cyano group, aldehyde group, and amide group were tolerated, and the reactions were highly regioselective, where in all cases 4-arylcoumarins were not observed. The crystallization of compound 3j from EtOAc gave a single crystal suitable for X-ray analysis. It illustrates the molecular structure of the substituted 3-arylcoumarin 3j (Fig. 1). Moreover, arylboronic acids with electron-donating groups (–CH3, –C(CH3)3, –OCH3, etc.) could promote the coupling reaction, and give better yields than those with electron-withdrawing groups (–F, –Cl, –Br, –CF3, –COCH3, –CN, –CHO, etc.). Especially, 3,4,5-trimethoxy phenylboronic acid could react with coumarin, giving 94% yield (Table 2 and 3e), where an almost quantitative yield was established. In addition, ortho-methoxy phenylboronic acid and α-naphthaleneboronic acid underwent smoothly this coupling reaction to generate the corresponding products 3f and 3g in good yield. The fact showed that the steric hindrance of arylboronic acids did not obviously affect this transformation. Gratifying, aliphatic boric acid, isopropyl boric acid could also react with coumarin to obtain 3-isopropylcoumarin 3zz. Unfortunately, benzyl acid failed to deliver the desired products with the current reaction system. Various substituted coumarins were also found to be amenable to this direct C–H functionalization reaction. Arylation of coumarins bearing alkyl and alkoxy proceeded smoothly leading to 3-arylcoumarins scaffolds 3s, 3u, and 3w in 72–93% yields. The highest yield 93% was obtained in transformation of 4-methyl-7-ethyloxycoumarin to its related product 3w with an almost quantitative yield. We were pleased to see that even sensitive functionalities such as hydroxyl and amino groups were also tolerated and the coupling reactions proceeded with no requisite for protection of these groups (3t, 3x and 3y). This feature is ubiquitous in hydroxycoumarin and aminocoumarin based biologically active products, which eliminates the requirement of protection and deprotection of hydroxyl and amino groups. Unfortunately, coumarin possessing an electron-withdrawing group such as –NO2 at the C6 position gave the desired product 3v in poor yield. It is worthy of note that these standard reaction conditions were also applied to 4-substituted coumarins, affording the corresponding products 3w, 3x and 3y in 93%, 48% and 49% yields, respectively. Moreover, the reaction of 3-acetyl coumarin with phenylboronic acid led to the formation of 3-phenylcoumarin derivative 3z with moderate yield (44%). These results indicated the steric hindrance of coumarins played a weak role in this reaction.
|
|---|
| a Reaction conditions: a solution of KMnO4 (1.0 mmol, 158 mg) in 20 mL AcOH was stirred under reflux until the purple color of KMnO4 turn brown (20 min). After the reaction was cooled to room temperature, coumarins 1 (0.5 mmol) and arylboronic acid 2 (1.0 mmol) were added and the reaction was continued at 80 °C for 0.5 h.b Isolated yields. |
 |

 | ||
| Fig. 1 X-ray crystal structure of 3j. | ||
In order to further explore the generality of this procedure, a series of quinolinone derivatives was also investigated under the optimal conditions (Table 3). We were pleased to observe that the C3 position of quinolinone derivatives were exclusively arylated, affording 3-arylquinolinones in 60–75% yields. Notably, N-methyl quinolinones with electron-rich (OCH3) and electron-poor (Br) groups on the phenyl ring were tolerated under these coupling conditions in good yield. Quinolinone with a bromine substituent also underwent the arylation reaction and resulted in 5d with an intact halo group to serve as a good precursor for further functionalizations. It was noteworthy that 2-quinolinone with a sensitive amide group was also tolerated and the coupling reaction proceeded with no requisite for protection of this group (5e).
|
|---|
| a Reaction conditions: a solution of KMnO4 (1.0 mmol, 158 mg) in 20 mL AcOH was stirred under reflux until the purple color of KMnO4 turn brown (20 min). After the reaction was cooled to room temperature, quinolinones 4 (0.5 mmol) and arylboronic acid 2 (1.0 mmol) were added and the reaction was continued at 80 °C for 0.5 h.b Isolated yields. |
 |

To investigate the reaction mechanism, some control experiments were conducted (Scheme 3). A <5% yield of 3a was obtained in the presence of the radical inhibitor (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) (eqn (1)), and a trace yield of 3a was produced when the radical scavenger butylated hydroxytoluene (BHT) was added (eqn (2)). These results could indicate that the reaction might proceed via a radical pathway. On the basis of these data and previous studies,19b,21 a possible reaction mechanism for the current manganese(III)-mediated C3-position direct radical arylation of coumarins was proposed as shown in Scheme 4. The formation of Mn(III) species could be explained via the reaction of KMnO4 with HOAc.21 The reaction of boronic acid A with Mn(III) salt generates aryl or alkyl radical B,19b,20b which attacks selectively C3-position of coumarin to give the carbon radical C stabilized by the conjugation with phenyl group. Subsequently, a single-electron transfer (SET) from C to Mn(III) would release the intermediate D, simultaneously Mn(III) was reduced into Mn(II). After that, the intermediate D loses a proton to produce the C3-functionalized coumarin E.
 | ||
| Scheme 3 Mechanistic investigations of the dehydrogenative radical coupling reaction. | ||
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have successfully developed a versatile and regioselective arylation of coumarins and quinolinones with arylboronic acids. This protocol provides a valuable approach to synthesize of biologically interesting 3-arylcoumarins via KMnO4/AcOH-mediated dehydrogenative direct radical coupling reaction. This reaction is high efficiency, moderate to good yield, and a broad functional groups tolerance.Experimental
General information
Anhydrous solvents were obtained by standard procedure. All substrates purchased from J & K Scientific Ltd. were used without further purification. Column chromatography was performed using 200–300 mesh silica with the indicated solvent system according to standard techniques. Analytical thin-layer chromatography (TLC) was performed on precoated, glass-backed silica gel plates. Visualization of the developed chromatogram was performed by UV absorbance (254 nm or 365 nm). Nuclear magnetic resonance spectra were recorded on Bruker Avance 400 MHz spectrometer. Chemical shifts for 1H NMR spectra are recorded in parts per million from tetramethylsilane. Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet and br = broad), coupling constant in Hz and integration. Chemical shifts for 13C NMR spectra were recorded in parts per million from tetramethylsilane. High resolution mass spectra (HR MS) were obtained on Q-TOF instrument using the ESI technique. IR spectra were recorded on Shimadazu IR-408 Fourier transform infrared spectrophotometer using a thin film supported on KBr pellets. Melting points were measured on an XT4A microscopic apparatus uncorrected.General procedure for synthesis of 3-aryl coumarin derivatives 3 (3-aryl quinolinone derivatives 5)
In a 50 mL Schlenk tube, a solution of KMnO4 (1.0 mmol, 158 mg) in 20 mL AcOH was stirred under reflux until the purple color of KMnO4 turn brown (20 min). After the reaction was cooled to room temperature, coumarins 1 (or quinolin derivatives 4) (0.5 mmol) and arylboronic acid 2 (1.0 mmol) were added and the reaction was continued at 80 °C for 0.5 h (monitored by TLC). The reaction mixture was diluted with EtOAc, and neutralized with the saturated NaHCO3. The resulting organic phase was dried over anhydrous NaSO4 and concentrated under vacuum. The crude product was purified by silica gel column chromatography using ethyl acetate/petroleum ether (1:5 to 2:1) as eluant to obtain the desired product 3 (or 5).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1601, 1454 (Ar–), 1117 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.71–7.69 (m, 2H), 7.53 (t, JH–H = 8.0 Hz, 2H), 7.47–7.40 (m, 3H), 7.36 (d, JH–H = 8.0 Hz, 3H), 7.29 (td, JH–H = 7.5 Hz, JH–H = 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.6, 153.5, 139.9 (CH), 134.7, 131.4 (CH), 128.8 (CH), 128.6 (CH), 128.5 (CH), 128.3, 127.9 (CH), 124.5 (CH), 119.7, 116.4 (CH). MS (ESI) m/z: 223.2 [M + H]+ (calcd for C15H11O2+ 223.0).O), 1610, 1452 (Ar–), 1113 (C–O). 1H NMR (400 MHz, DMSO) δ: 8.19 (s, 1H), 7.75 (d, JH–H = 7.6 Hz, 1H), 7.63–7.58 (m, 3H), 7.41 (d, JH–H = 8.2 Hz, 1H), 7.36 (t, JH–H = 7.5 Hz, 1H), 7.25 (d, JH–H = 8.0 Hz, 2H), 2.34 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 160.2, 153.2, 140.3 (CH), 138.5, 132.1, 131.9 (CH), 129.2 (CH), 128.9 (CH), 128.7 (CH), 127.1, 125.0 (CH), 119.9, 116.2 (CH), 21.2 (CH3). MS (ESI) m/z: 237.3 [M + H]+ (calcd for C16H13O2+ 237.0).O), 1603, 1450 (Ar–), 1124 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.78 (s, 1H), 7.65 (d, JH–H = 8.5 Hz, 1H), 7.53–7.50 (m, 2H), 7.46 (d, JH–H = 8.5 Hz, 2H), 7.34 (d, JH–H = 8.0 Hz, 1H), 7.27 (td, JH–H = 7.5 Hz, JH–H = 1.0 Hz, 1H), 1.34 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 160.7, 153.4, 152.0, 139.3 (CH), 131.8, 131.2 (CH), 128.3, 128.2 (CH), 127.8 (CH), 125.4 (CH), 124.4 (CH), 119.7, 116.4 (CH), 34.7, 31.2 (CH3). MS (ESI) m/z: 279.2 [M + H]+ (calcd for C19H19O2+ 279.1).O), 1608, 1514, 1452 (Ar–), 1252, 1128 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.74 (s, 1H), 7.66 (d, JH–H = 8.9 Hz, 2H), 7.52–7.46 (m, 2H), 7.33 (d, JH–H = 8.2 Hz, 1H), 7.27 (td, JH–H = 7.5 Hz, JH–H = 1.1 Hz, 1H), 6.96 (d, JH–H = 8.9 Hz, 2H), 2.83 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.8, 160.1, 153.2, 138.5 (CH), 131.0 (CH), 129.8 (CH), 127.8, 127.7 (CH), 127.1, 124.4 (CH), 119.8, 116.3 (CH), 113.9 (CH), 55.3 (CH3). MS (ESI) m/z: 253.4 [M + H]+ (calcd for C16H13O3+ 253.0).O), 1606, 1588, 1508, 1450 (Ar–), 1242, 1126 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.56–7.50 (m, 2H), 7.33 (d, JH–H = 8.2 Hz, 1H), 7.29 (td, JH–H = 7.4 Hz, JH–H = 1.0 Hz, 1H), 6.94 (s, 2H), 3.91 (s, 6H), 3.89 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.5, 153.3, 153.0, 139.5 (CH), 138.7, 131.4 (CH), 130.1, 128.0, 127.9 (CH), 124.5 (CH), 119.5, 116.3 (CH), 106.0 (CH), 60.8 (CH3), 56.2 (CH3). MS (ESI) m/z: 313.0 [M + H]+ (calcd for C18H17O5+ 313.1).O), 1608, 1491, 1456 (Ar–), 1246, 1130 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.73 (s, 1H), 7.49 (d, JH–H = 7.4 Hz, 2H), 7.38–7.34 (m, 3H), 7.27 (t, JH–H = 7.4 Hz, 1H), 7.02 (t, JH–H = 7.5 Hz, 1H), 6.99 (d, JH–H = 8.2 Hz, 1H), 3.82 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.2, 157.3, 153.7, 141.7 (CH), 131.1 (CH), 130.7 (CH), 130.2 (CH), 127.8 (CH), 126.6, 124.2 (CH), 124.1, 120.6 (CH), 119.5, 116.5 (CH), 111.4 (CH), 55.7 (CH3). MS (ESI) m/z: 253.2 [M + H]+ (calcd for C16H13O3+ 253.1).O), 1606, 1454 (Ar–), 1132 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.89 (td, JH–H = 6.8 Hz, JH–H = 1.9 Hz, 2H), 7.78 (s, 1H), 7.77 (dd, JH–H = 8.2 Hz, JH–H = 1.3 Hz, 1H), 7.56–7.46 (m, 6H), 7.43 (t, JH–H = 8.5 Hz, 1H), 7.30 (td, JH–H = 7.6 Hz, JH–H = 1.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.8, 154.0, 142.8 (CH), 133.7, 132.7, 131.7 (CH), 131.6, 129.4 (CH), 128.6 (CH), 128.3, 128.0 (CH), 127.7 (CH), 126.5 (CH), 126.1 (CH), 125.3 (CH), 125.1 (CH), 124.6 (CH), 119.3, 116.7 (CH). MS (ESI) m/z: 273.2 [M + H]+ (calcd for C19H13O2+ 273.1).O), 1604, 1514, 1454 (Ar–), 1236 (C–F), 1128 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.79 (s, 1H), 7.71–7.68 (m, 2H), 7.53 (td, JH–H = 7.5 Hz, JH–H = 1.4 Hz, 2H), 7.36 (d, JH–H = 8.1 Hz, 1H), 7.30 (td, JH–H = 8.5 Hz, JH–H = 1.0 Hz, 1H), 7.15–7.11 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 163.1 (d, JF–C = 247.3 Hz), 160.5, 153.4, 139.7 (CH), 131.5 (CH), 130.7 (d, JF–C = 3.1 Hz), 130.4 (d, JF–C = 8.1 Hz, CH), 127.9 (CH), 127.3, 124.6 (CH), 119.5, 116.5 (CH), 115.5 (d, JF–C = 3.1 Hz, CH). 19F NMR (376 MHz, CDCl3) δ: −112.3. MS (ESI) m/z: 241.1 [M + H]+ (calcd for C15H10FO2+ 241.0).O), 1608, 1489, 1452 (Ar–), 1098 (C–O), 748 (C–Cl). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.65 (d, JH–H = 8.5 Hz, 2H), 7.55–7.51 (m, 2H), 7.40 (d, JH–H = 8.6 Hz, 2H), 7.35 (d, JH–H = 8.6 Hz, 1H), 7.30 (td, JH–H = 8.5 Hz, JH–H = 0.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.4, 153.5, 139.9 (CH), 134.9, 133.0, 131.7 (CH), 129.8 (CH), 128.7 (CH), 128.0 (CH), 127.1, 124.6 (CH), 119.5, 116.5 (CH). MS (ESI) m/z: 257.3 [M + H]+ (calcd for C15H10ClO2+ 257.0).O), 1610, 1487, 1450 (Ar–), 1012 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.60–7.52 (m, 6H), 7.35 (d, JH–H = 8.7 Hz, 1H), 7.30 (td, JH–H = 8.6 Hz, JH–H = 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.2, 153.5, 139.9 (CH), 133.5, 131.7 (CH), 131.6 (CH), 130.1 (CH), 128.0 (CH), 127.1, 124.6 (CH), 123.1, 119.5, 116.5 (CH). MS (ESI) m/z: 301.1 [M + H]+ (calcd for C15H10BrO2+ 301.0).O), 1610, 1588, 1456 (Ar–), 1180 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.83 (s, 1H), 7.54 (t, JH–H = 7.4 Hz, 2H), 7.47 (t, JH–H = 7.5 Hz, 2H), 7.43–7.39 (m, 1H), 7.36 (d, JH–H = 8.6 Hz, 1H), 7.30 (d, JH–H = 7.9 Hz, 1H), 7.09 (td, JH–H = 8.6 Hz, JH–H = 1.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 162.6 (d, JF–C = 244.4 Hz), 160.2, 153.5, 140.4 (CH), 136.6 (d, JF–C = 8.2 Hz), 131.8 (CH), 130.0 (d, JF–C = 8.2 Hz, CH), 128.1 (CH), 127.0 (d, JF–C = 2.2 Hz, CH), 124.6 (CH), 124.1 (d, JF–C = 2.9 Hz, CH), 119.4, 116.5 (CH), 115.8 (d, JF–C = 8.8 Hz, CH), 115.6 (d, JF–C = 10.7 Hz, CH). 19F NMR (376 MHz, CDCl3) δ: −112.6. HR MS (ESI) m/z: 241.0655 [M + H]+ (calcd for C15H10FO2+ 241.0659).O), 1608, 1456, 1356 (Ar–), 1101 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.88 (s, 1H), 7.83 (d, JH–H = 8.1 Hz, 2H), 7.70 (d, JH–H = 8.2 Hz, 2H), 7.59–7.55 (m, 2H), 7.38 (d, JH–H = 8.7 Hz, 1H), 7.33 (td, JH–H = 8.5 Hz, JH–H = 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.2, 153.7, 140.9 (CH), 138.2, 132.1 (CH), 130.7 (d, JF–C = 31.2 Hz), 128.9 (CH), 128.2 (CH), 128.0, 126.9, 125.4 (d, JF–C = 3.7 Hz, CH), 124.7 (CH), 119.3, 116.6 (CH). 19F NMR (376 MHz, CDCl3) δ: −62.7. MS (ESI) m/z: 291.2 [M + H]+ (calcd for C16H10F3O2+ 291.0).O), 1604, 1454 (Ar–), 1109 (C–O). 1H NMR (400 MHz, CDCl3) δ: 8.11 (dd, JH–H = 8.4 Hz, JH–H = 1.8 Hz, 2H), 7.89 (s, 1H), 7.80 (dd, JH–H = 8.3 Hz, JH–H = 1.8 Hz, 2H), 7.57 (td, JH–H = 7.5 Hz, JH–H = 1.6 Hz, 2H), 7.39 (d, JH–H = 8.6 Hz, 1H), 7.32 (t, JH–H = 7.1 Hz, 1H), 3.94 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 166.6, 160.1, 153.7, 140.8 (CH), 139.1, 131.9 (CH), 130.2, 129.7 (CH), 128.5 (CH), 128.1 (CH), 127.3, 124.7 (CH), 119.4, 116.5 (CH), 52.2 (CH3). MS (ESI) m/z: 281.3 [M + H]+ (calcd for C17H13O4+ 281.1).O), 1604, 1574, 1454 (Ar–), 1111 (C–O). 1H NMR (400 MHz, CDCl3) δ: 8.02 (d, JH–H = 8.2 Hz, 2H), 7.90 (s, 1H), 7.82 (d, JH–H = 8.2 Hz, 2H), 7.57 (t, JH–H = 7.6 Hz, 2H), 7.38 (d, JH–H = 8.2 Hz, 1H), 7.32 (t, JH–H = 7.5 Hz, 1H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 197.5, 160.1, 153.7, 140.8 (CH), 139.2, 137.0, 132.0 (CH), 128.7 (CH), 128.4, 128.1 (CH), 127.1, 124.7 (CH), 119.4, 116.5 (CH), 26.7 (CH3). MS (ESI) m/z: 265.3 [M + H]+ (calcd for C17H13O3+ 265.1).O), 1606, 1473, 1452 (Ar–), 1028 (C–O), 752 (C–Cl). 1H NMR (400 MHz, CDCl3) δ: 7.84–7.83 (m, 2H), 7.61–7.51 (m, 4H), 7.38 (d, JH–H = 8.6 Hz, 1H), 7.32 (td, JH–H = 7.7 Hz, JH–H = 0.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.0, 153.6, 140.4 (CH), 134.5, 133.1, 132.7, 132.0 (CH), 130.4 (CH), 130.3 (CH), 128.1 (CH), 127.8 (CH), 125.9, 124.7 (CH), 119.2, 116.6 (CH). MS (ESI) m/z: 291.2 [M + H]+ (calcd for C15H9Cl2O2+ 291.0).O), 1614, 1446 (Ar–), 1115 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.90 (s, 1H), 7.85 (d, JH–H = 8.4 Hz, 2H), 7.74 (d, JH–H = 8.4 Hz, 2H), 7.59 (d, JH–H = 7.5 Hz, 2H), 7.39 (d, JH–H = 8.4 Hz, 1H), 7.34 (td, JH–H = 8.4 Hz, JH–H = 0.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 159.9, 153.7, 141.3 (CH), 139.1 (CH), 132.4 (CH), 132.2 (CH), 129.2 (CH), 128.3 (CH), 126.4, 124.8 (CH), 119.2, 118.5, 116.6 (CH), 112.4. MS (ESI) m/z: 248.2 [M + H]+ (calcd for C16H10NO2+ 248.0).O), 1611, 1434 (Ar–), 1113 (C–O). 1H NMR (400 MHz, DMSO) δ: 10.06 (s, 1H), 8.41 (s, 1H), 8.01–7.96 (m, 4H), 7.82 (d, JH–H = 7.5 Hz, 1H), 7.66 (t, JH–H = 7.5 Hz, 1H), 7.46 (d, JH–H = 8.3 Hz, 1H), 7.41 (t, JH–H = 7.5 Hz, 1H). 13C NMR (100 MHz, DMSO) δ: 193.2 (CHO), 159.8, 153.6, 142.5 (CH), 140.9, 136.2, 132.8 (CH), 129.7 (CH), 129.6 (CH), 129.4 (CH), 126.2, 125.2 (CH), 119.7, 116.4 (CH). HR MS (ESI) m/z: 251.0707 [M + H]+ (calcd for C16H11NO3+ 251.0703).O), 1660 (CO), 1605, 1430 (Ar–). 1H NMR (400 MHz, DMSO) δ: 10.08 (s, 1H), 8.17 (s, 1H), 7.93 (s, 1H), 7.76 (dd, JH–H = 7.7 Hz, JH–H = 1.1 Hz, 1H), 7.70 (dd, JH–H = 7.1 Hz, JH–H = 1.9 Hz, 1H), 7.60 (td, JH–H = 7.8 Hz, JH–H = 1.4 Hz, 1H), 7.42–7.33 (m, 4H). 13C NMR (100 MHz, DMSO) δ: 168.9, 160.0, 153.4, 140.9 (CH), 139.6, 135.4, 132.1 (CH), 129.0 (CH), 128.9 (CH), 127.2, 125.0 (CH), 123.6 (CH), 119.8, 119.7 (CH), 119.6 (CH), 116.2 (CH), 24.4 (CH3). HR MS (ESI) m/z: 280.0966 [M + H]+ (calcd for C17H14NO3+ 280.0968).O), 1616, 1577, 1448 (Ar–), 1111 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.74 (s, 1H), 7.70–7.67 (m, 2H), 7.46–7.37 (m, 3H), 7.33–7.31 (m, 2H), 7.24 (dd, JH–H = 7.4 Hz, JH–H = 1.6 Hz, 1H), 2.41 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.8, 151.6, 139.9 (CH), 134.8, 134.1, 132.4 (CH), 128.7 (CH), 128.5 (CH), 128.4 (CH), 128.2, 127.7 (CH), 119.4, 116.1 (CH), 20.8 (CH3). MS (ESI) m/z: 237.2 [M + H]+ (calcd for C16H13O2+ 237.1).O), 1615, 1595, 1570, 1445 (Ar–), 1080 (C–O). 1H NMR (400 MHz, DMSO) δ: 10.66 (s, 1H), 8.15 (s, 1H), 7.69 (d, JH–H = 7.3 Hz, 2H), 7.60 (d, JH–H = 8.5 Hz, 1H), 7.43 (d, JH–H = 7.6 Hz, 2H), 7.37 (t, JH–H = 7.3 Hz, 1H), 6.82 (dd, JH–H = 8.5 Hz, JH–H = 2.2 Hz, 1H), 6.76 (d, JH–H = 2.0 Hz, 1H). 13C NMR (100 MHz, DMSO) δ: 161.7, 160.6, 155.3, 141.6 (CH), 135.6, 130.4 (CH), 128.7 (CH), 128.6 (CH), 128.5 (CH), 122.6, 113.8 (CH), 112.4 (CH), 102.2 (CH). MS (ESI) m/z: 239.1 [M + H]+ (calcd for C15H11O3+ 239.0).O), 1614, 1506, 1464, 1439 (Ar–), 1120 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.75 (s, 1H), 7.68 (d, JH–H = 8.4 Hz, 2H), 7.45–7.41 (m, 3H), 7.39–7.35 (m, 1H), 6.87–6.84 (m, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 162.6, 160.8, 155.3, 139.9 (CH), 135.0, 128.8 (CH), 128.4 (CH), 124.8, 113.3, 112.7 (CH), 100.4 (CH), 55.7 (CH3). MS (ESI) m/z: 253.3 [M + H]+ (calcd for C16H13O3+ 253.1).O), 1604, 1508, 1385, 1361 (Ar–), 1074 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.52 (d, JH–H = 8.8 Hz, 1H), 7.41 (d, JH–H = 7.0 Hz, 2H), 7.35 (d, JH–H = 7.3 Hz, 1H), 7.29–7.25 (m, 2H), 6.85 (dd, JH–H = 8.8 Hz, JH–H = 2.5 Hz, 1H), 6.79 (d, JH–H = 2.2 Hz, 1H), 4.06 (q, JH–H = 7.0 Hz, 2H), 2.23 (s, 3H), 1.43 (t, JH–H = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.7, 161.3, 154.3, 148.0, 134.7, 130.2 (CH), 128.3 (CH), 127.9 (CH), 126.1 (CH), 124.0, 113.9, 112.6 (CH), 101.0 (CH), 64.1 (CH2), 16.5 (CH3), 14.6 (CH3). HR MS (ESI) m/z: 281.1175 [M + H]+ (calcd for C18H17O3+ 281.1172).O), 1614, 1593, 1577, 1446 (Ar–), 1078 (C–O). 1H NMR (400 MHz, DMSO) δ: 10.5 (s, –OH), 7.61 (d, JH–H = 8.8 Hz, 1H), 7.43 (t, JH–H = 7.5 Hz, 2H), 7.36 (t, JH–H = 7.2 Hz, 1H), 7.28 (d, JH–H = 6.9 Hz, 2H), 6.83 (dd, JH–H = 8.8 Hz, JH–H = 2.2 Hz, 1H), 6.74 (d, JH–H = 2.2 Hz, 1H), 2.17 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 161.3, 160.7, 154.3, 148.7, 135.3, 130.7 (CH), 128.4 (CH), 128.0 (CH), 127.5 (CH), 122.7, 113.4 (CH), 112.8, 102.3 (CH), 16.7 (CH3). MS (ESI) m/z: 253.2 [M + H]+ (calcd for C16H13O3+ 253.1).O), 1613, 1445 (Ar–), 1117 (C–O). 1H NMR (400 MHz, DMSO) δ: 7.47 (d, JH–H = 8.7 Hz, 1H), 7.42 (t, JH–H = 7.5 Hz, 2H), 7.35 (t, JH–H = 7.3 Hz, 1H), 7.25 (d, JH–H = 7.0 Hz, 2H), 6.60 (dd, JH–H = 8.7 Hz, JH–H = 2.0 Hz, 1H), 6.46 (d, JH–H = 2.0 Hz, 1H), 6.13 (bs, 2H), 2.14 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 161.1, 154.9, 153.2, 149.1, 135.8, 130.9 (CH), 128.4 (CH), 127.7 (CH), 127.2 (CH), 120.1, 111.8 (CH), 109.5, 98.7 (CH), 16.6 (CH3). MS (ESI) m/z: 252.3 [M + H]+ (calcd for C16H14NO2+ 252.1).O), 1608, 1562, 1448, 1363 (Ar–), 1049 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.60–7.56 (m, 1H), 7.52–7.50 (m, 3H), 7.40 (d, JH–H = 8.3 Hz, 1H), 7.32–7.30 (m, 2H), 7.22 (d, JH–H = 4.1 Hz, 1H), 2.24 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 199.0, 158.4, 153.4, 151.8, 132.7 (CH), 132.5, 129.6 (CH), 128.8 (CH), 128.5 (CH), 128.1 (CH), 127.7, 124.6 (CH), 119.4 (CH), 117.0 (CH), 31.1 (CH3). MS (ESI) m/z: 265.1 [M + H]+ (calcd for C17H13O3+ 265.0).O), 1610, 1452 (Ar–), 1387, 1190 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.47–7.44 (m, 3H), 7.30 (d, JH–H = 8.6 Hz, 1H), 7.25 (t, JH–H = 7.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 161.4, 152.8, 135.9 (CH), 135.7, 130.4 (CH), 127.2 (CH), 124.1 (CH), 119.5, 116.3 (CH), 28.7 (CH), 21.4 (CH3). MS (ESI) m/z: 189.1 [M + H]+ (calcd for C12H13O2+ 189.0).O), 1591, 1454 (Ar–). 1H NMR (400 MHz, CDCl3) δ: 7.77 (s, 1H), 7.70 (dd, JH–H = 7.0 Hz, JH–H = 1.4 Hz, 2H), 7.59–7.25 (m, 2H), 7.42 (t, JH–H = 7.5 Hz, 2H), 7.35 (t, JH–H = 8.4 Hz, 2H), 7.22 (t, JH–H = 7.3 Hz, 1H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.5, 139.6, 136.8 (CH), 132.4, 130.3 (CH), 128.9 (CH), 128.8 (CH), 128.1 (CH), 128.0 (CH), 122.2 (CH), 120.7, 114.0 (CH), 29.9 (CH3). MS (ESI) m/z: 236.2 [M + H]+ (calcd for C16H14NO+ 236.1).O), 1604, 1598, 1510, 1458 (Ar–), 1246, 1178, 1030. 1H NMR (400 MHz, CDCl3) δ: 7.73 (s, 1H), 7.67 (dd, JH–H = 7.0 Hz, JH–H = 1.9 Hz, 2H), 7.56 (dd, JH–H = 7.8 Hz, JH–H = 1.0 Hz, 1H), 7.51 (td, JH–H = 7.2 Hz, JH–H = 1.4 Hz, 1H), 7.32 (d, JH–H = 8.5 Hz, 1H), 7.21 (td, JH–H = 7.2 Hz, JH–H = 0.6 Hz, 1H), 6.95 (d, JH–H = 8.8 Hz, 2H), 3.83 (s, 3H), 3.76 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.6, 159.5, 139.3, 135.7 (CH), 131.9, 130.2 (CH), 129.9 (CH), 129.2, 128.6 (CH), 122.1 (CH), 120.8, 113.9 (CH), 113.6 (CH), 55.3 (CH3), 29.9 (CH3). MS (ESI) m/z: 266.2 [M + H]+ (calcd for C17H16NO2+ 266.1).O), 1616, 1595, 1508, 1454 (Ar–), 1248, 1211, 1036. 1H NMR (400 MHz, CDCl3) δ: 7.70–7.67 (m, 3H), 7.48 (d, JH–H = 8.6 Hz, 1H), 7.40 (td, JH–H = 7.2 Hz, JH–H = 1.2 Hz, 2H), 7.35–7.31 (m, 1H), 6.81 (dd, JH–H = 8.6 Hz, JH–H = 2.2 Hz, 1H), 6.76 (d, JH–H = 2.2 Hz, 1H), 3.90 (s, 3H), 3.72 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.8, 161.6, 141.2, 137.0, 136.7 (CH), 130.2 (CH), 129.2, 128.8 (CH), 128.1 (CH), 127.7 (CH), 114.9, 109.8 (CH), 98.4 (CH), 55.6 (CH3), 29.9 (CH3). HR MS (ESI) m/z: 266.1179 [M + H]+ (calcd for C17H16NO2+ 266.1176).O), 1579, 1487, 1444, 1415 (Ar–), 1228, 1209, 1118. 1H NMR (400 MHz, CDCl3) δ: 7.73 (d, JH–H = 2.2 Hz, 1H), 7.70–7.67 (m, 3H), 7.63 (dd, JH–H = 8.9 Hz, JH–H = 2.2 Hz, 1H), 7.44 (t, JH–H = 7.6 Hz, 2H), 7.40–7.37 (m, 1H), 7.25 (d, JH–H = 6.7 Hz, 1H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.1, 138.5, 136.3, 135.3 (CH), 133.7, 132.9 (CH), 130.8 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 122.2, 115.7 (CH), 114.9, 30.1 (CH3). MS (ESI) m/z: 314.1 [M + H]+ (calcd for C16H13BrNO+ 314.0).O), 1610, 1464, 1454 (Ar–), 1120, 1028. 1H NMR (400 MHz, DMSO) δ: 11.96 (s, 1H), 8.10 (s, 1H), 7.77–7.72 (m, 3H), 7.50 (td, JH–H = 8.3 Hz, JH–H = 1.2 Hz, 1H), 7.45–7.41 (m, 2H), 7.40 (d, JH–H = 7.3 Hz, 1H), 7.34 (d, JH–H = 8.1 Hz, 1H), 7.20 (td, JH–H = 7.9 Hz, JH–H = 0.8 Hz, 1H). 13C NMR (100 MHz, DMSO) δ: 161.5, 138.8, 138.0 (CH), 136.7, 131.9, 130.6 (CH), 129.1 (CH), 128.6 (CH), 128.4 (CH), 128.3 (CH), 122.3 (CH), 120.0, 115.1 (CH). MS (ESI) m/z: 222.3 [M + H]+ (calcd for C15H12NO+ 222.1).
O), 1601, 1454 (Ar–), 1117 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.71–7.69 (m, 2H), 7.53 (t, JH–H = 8.0 Hz, 2H), 7.47–7.40 (m, 3H), 7.36 (d, JH–H = 8.0 Hz, 3H), 7.29 (td, JH–H = 7.5 Hz, JH–H = 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.6, 153.5, 139.9 (CH), 134.7, 131.4 (CH), 128.8 (CH), 128.6 (CH), 128.5 (CH), 128.3, 127.9 (CH), 124.5 (CH), 119.7, 116.4 (CH). MS (ESI) m/z: 223.2 [M + H]+ (calcd for C15H11O2+ 223.0).O), 1610, 1452 (Ar–), 1113 (C–O). 1H NMR (400 MHz, DMSO) δ: 8.19 (s, 1H), 7.75 (d, JH–H = 7.6 Hz, 1H), 7.63–7.58 (m, 3H), 7.41 (d, JH–H = 8.2 Hz, 1H), 7.36 (t, JH–H = 7.5 Hz, 1H), 7.25 (d, JH–H = 8.0 Hz, 2H), 2.34 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 160.2, 153.2, 140.3 (CH), 138.5, 132.1, 131.9 (CH), 129.2 (CH), 128.9 (CH), 128.7 (CH), 127.1, 125.0 (CH), 119.9, 116.2 (CH), 21.2 (CH3). MS (ESI) m/z: 237.3 [M + H]+ (calcd for C16H13O2+ 237.0).O), 1603, 1450 (Ar–), 1124 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.78 (s, 1H), 7.65 (d, JH–H = 8.5 Hz, 1H), 7.53–7.50 (m, 2H), 7.46 (d, JH–H = 8.5 Hz, 2H), 7.34 (d, JH–H = 8.0 Hz, 1H), 7.27 (td, JH–H = 7.5 Hz, JH–H = 1.0 Hz, 1H), 1.34 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 160.7, 153.4, 152.0, 139.3 (CH), 131.8, 131.2 (CH), 128.3, 128.2 (CH), 127.8 (CH), 125.4 (CH), 124.4 (CH), 119.7, 116.4 (CH), 34.7, 31.2 (CH3). MS (ESI) m/z: 279.2 [M + H]+ (calcd for C19H19O2+ 279.1).O), 1608, 1514, 1452 (Ar–), 1252, 1128 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.74 (s, 1H), 7.66 (d, JH–H = 8.9 Hz, 2H), 7.52–7.46 (m, 2H), 7.33 (d, JH–H = 8.2 Hz, 1H), 7.27 (td, JH–H = 7.5 Hz, JH–H = 1.1 Hz, 1H), 6.96 (d, JH–H = 8.9 Hz, 2H), 2.83 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.8, 160.1, 153.2, 138.5 (CH), 131.0 (CH), 129.8 (CH), 127.8, 127.7 (CH), 127.1, 124.4 (CH), 119.8, 116.3 (CH), 113.9 (CH), 55.3 (CH3). MS (ESI) m/z: 253.4 [M + H]+ (calcd for C16H13O3+ 253.0).O), 1606, 1588, 1508, 1450 (Ar–), 1242, 1126 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.56–7.50 (m, 2H), 7.33 (d, JH–H = 8.2 Hz, 1H), 7.29 (td, JH–H = 7.4 Hz, JH–H = 1.0 Hz, 1H), 6.94 (s, 2H), 3.91 (s, 6H), 3.89 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.5, 153.3, 153.0, 139.5 (CH), 138.7, 131.4 (CH), 130.1, 128.0, 127.9 (CH), 124.5 (CH), 119.5, 116.3 (CH), 106.0 (CH), 60.8 (CH3), 56.2 (CH3). MS (ESI) m/z: 313.0 [M + H]+ (calcd for C18H17O5+ 313.1).O), 1608, 1491, 1456 (Ar–), 1246, 1130 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.73 (s, 1H), 7.49 (d, JH–H = 7.4 Hz, 2H), 7.38–7.34 (m, 3H), 7.27 (t, JH–H = 7.4 Hz, 1H), 7.02 (t, JH–H = 7.5 Hz, 1H), 6.99 (d, JH–H = 8.2 Hz, 1H), 3.82 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.2, 157.3, 153.7, 141.7 (CH), 131.1 (CH), 130.7 (CH), 130.2 (CH), 127.8 (CH), 126.6, 124.2 (CH), 124.1, 120.6 (CH), 119.5, 116.5 (CH), 111.4 (CH), 55.7 (CH3). MS (ESI) m/z: 253.2 [M + H]+ (calcd for C16H13O3+ 253.1).O), 1606, 1454 (Ar–), 1132 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.89 (td, JH–H = 6.8 Hz, JH–H = 1.9 Hz, 2H), 7.78 (s, 1H), 7.77 (dd, JH–H = 8.2 Hz, JH–H = 1.3 Hz, 1H), 7.56–7.46 (m, 6H), 7.43 (t, JH–H = 8.5 Hz, 1H), 7.30 (td, JH–H = 7.6 Hz, JH–H = 1.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.8, 154.0, 142.8 (CH), 133.7, 132.7, 131.7 (CH), 131.6, 129.4 (CH), 128.6 (CH), 128.3, 128.0 (CH), 127.7 (CH), 126.5 (CH), 126.1 (CH), 125.3 (CH), 125.1 (CH), 124.6 (CH), 119.3, 116.7 (CH). MS (ESI) m/z: 273.2 [M + H]+ (calcd for C19H13O2+ 273.1).O), 1604, 1514, 1454 (Ar–), 1236 (C–F), 1128 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.79 (s, 1H), 7.71–7.68 (m, 2H), 7.53 (td, JH–H = 7.5 Hz, JH–H = 1.4 Hz, 2H), 7.36 (d, JH–H = 8.1 Hz, 1H), 7.30 (td, JH–H = 8.5 Hz, JH–H = 1.0 Hz, 1H), 7.15–7.11 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 163.1 (d, JF–C = 247.3 Hz), 160.5, 153.4, 139.7 (CH), 131.5 (CH), 130.7 (d, JF–C = 3.1 Hz), 130.4 (d, JF–C = 8.1 Hz, CH), 127.9 (CH), 127.3, 124.6 (CH), 119.5, 116.5 (CH), 115.5 (d, JF–C = 3.1 Hz, CH). 19F NMR (376 MHz, CDCl3) δ: −112.3. MS (ESI) m/z: 241.1 [M + H]+ (calcd for C15H10FO2+ 241.0).O), 1608, 1489, 1452 (Ar–), 1098 (C–O), 748 (C–Cl). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.65 (d, JH–H = 8.5 Hz, 2H), 7.55–7.51 (m, 2H), 7.40 (d, JH–H = 8.6 Hz, 2H), 7.35 (d, JH–H = 8.6 Hz, 1H), 7.30 (td, JH–H = 8.5 Hz, JH–H = 0.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.4, 153.5, 139.9 (CH), 134.9, 133.0, 131.7 (CH), 129.8 (CH), 128.7 (CH), 128.0 (CH), 127.1, 124.6 (CH), 119.5, 116.5 (CH). MS (ESI) m/z: 257.3 [M + H]+ (calcd for C15H10ClO2+ 257.0).O), 1610, 1487, 1450 (Ar–), 1012 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.81 (s, 1H), 7.60–7.52 (m, 6H), 7.35 (d, JH–H = 8.7 Hz, 1H), 7.30 (td, JH–H = 8.6 Hz, JH–H = 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.2, 153.5, 139.9 (CH), 133.5, 131.7 (CH), 131.6 (CH), 130.1 (CH), 128.0 (CH), 127.1, 124.6 (CH), 123.1, 119.5, 116.5 (CH). MS (ESI) m/z: 301.1 [M + H]+ (calcd for C15H10BrO2+ 301.0).O), 1610, 1588, 1456 (Ar–), 1180 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.83 (s, 1H), 7.54 (t, JH–H = 7.4 Hz, 2H), 7.47 (t, JH–H = 7.5 Hz, 2H), 7.43–7.39 (m, 1H), 7.36 (d, JH–H = 8.6 Hz, 1H), 7.30 (d, JH–H = 7.9 Hz, 1H), 7.09 (td, JH–H = 8.6 Hz, JH–H = 1.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 162.6 (d, JF–C = 244.4 Hz), 160.2, 153.5, 140.4 (CH), 136.6 (d, JF–C = 8.2 Hz), 131.8 (CH), 130.0 (d, JF–C = 8.2 Hz, CH), 128.1 (CH), 127.0 (d, JF–C = 2.2 Hz, CH), 124.6 (CH), 124.1 (d, JF–C = 2.9 Hz, CH), 119.4, 116.5 (CH), 115.8 (d, JF–C = 8.8 Hz, CH), 115.6 (d, JF–C = 10.7 Hz, CH). 19F NMR (376 MHz, CDCl3) δ: −112.6. HR MS (ESI) m/z: 241.0655 [M + H]+ (calcd for C15H10FO2+ 241.0659).O), 1608, 1456, 1356 (Ar–), 1101 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.88 (s, 1H), 7.83 (d, JH–H = 8.1 Hz, 2H), 7.70 (d, JH–H = 8.2 Hz, 2H), 7.59–7.55 (m, 2H), 7.38 (d, JH–H = 8.7 Hz, 1H), 7.33 (td, JH–H = 8.5 Hz, JH–H = 1.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.2, 153.7, 140.9 (CH), 138.2, 132.1 (CH), 130.7 (d, JF–C = 31.2 Hz), 128.9 (CH), 128.2 (CH), 128.0, 126.9, 125.4 (d, JF–C = 3.7 Hz, CH), 124.7 (CH), 119.3, 116.6 (CH). 19F NMR (376 MHz, CDCl3) δ: −62.7. MS (ESI) m/z: 291.2 [M + H]+ (calcd for C16H10F3O2+ 291.0).O), 1604, 1454 (Ar–), 1109 (C–O). 1H NMR (400 MHz, CDCl3) δ: 8.11 (dd, JH–H = 8.4 Hz, JH–H = 1.8 Hz, 2H), 7.89 (s, 1H), 7.80 (dd, JH–H = 8.3 Hz, JH–H = 1.8 Hz, 2H), 7.57 (td, JH–H = 7.5 Hz, JH–H = 1.6 Hz, 2H), 7.39 (d, JH–H = 8.6 Hz, 1H), 7.32 (t, JH–H = 7.1 Hz, 1H), 3.94 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 166.6, 160.1, 153.7, 140.8 (CH), 139.1, 131.9 (CH), 130.2, 129.7 (CH), 128.5 (CH), 128.1 (CH), 127.3, 124.7 (CH), 119.4, 116.5 (CH), 52.2 (CH3). MS (ESI) m/z: 281.3 [M + H]+ (calcd for C17H13O4+ 281.1).O), 1604, 1574, 1454 (Ar–), 1111 (C–O). 1H NMR (400 MHz, CDCl3) δ: 8.02 (d, JH–H = 8.2 Hz, 2H), 7.90 (s, 1H), 7.82 (d, JH–H = 8.2 Hz, 2H), 7.57 (t, JH–H = 7.6 Hz, 2H), 7.38 (d, JH–H = 8.2 Hz, 1H), 7.32 (t, JH–H = 7.5 Hz, 1H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 197.5, 160.1, 153.7, 140.8 (CH), 139.2, 137.0, 132.0 (CH), 128.7 (CH), 128.4, 128.1 (CH), 127.1, 124.7 (CH), 119.4, 116.5 (CH), 26.7 (CH3). MS (ESI) m/z: 265.3 [M + H]+ (calcd for C17H13O3+ 265.1).O), 1606, 1473, 1452 (Ar–), 1028 (C–O), 752 (C–Cl). 1H NMR (400 MHz, CDCl3) δ: 7.84–7.83 (m, 2H), 7.61–7.51 (m, 4H), 7.38 (d, JH–H = 8.6 Hz, 1H), 7.32 (td, JH–H = 7.7 Hz, JH–H = 0.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 160.0, 153.6, 140.4 (CH), 134.5, 133.1, 132.7, 132.0 (CH), 130.4 (CH), 130.3 (CH), 128.1 (CH), 127.8 (CH), 125.9, 124.7 (CH), 119.2, 116.6 (CH). MS (ESI) m/z: 291.2 [M + H]+ (calcd for C15H9Cl2O2+ 291.0).O), 1614, 1446 (Ar–), 1115 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.90 (s, 1H), 7.85 (d, JH–H = 8.4 Hz, 2H), 7.74 (d, JH–H = 8.4 Hz, 2H), 7.59 (d, JH–H = 7.5 Hz, 2H), 7.39 (d, JH–H = 8.4 Hz, 1H), 7.34 (td, JH–H = 8.4 Hz, JH–H = 0.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 159.9, 153.7, 141.3 (CH), 139.1 (CH), 132.4 (CH), 132.2 (CH), 129.2 (CH), 128.3 (CH), 126.4, 124.8 (CH), 119.2, 118.5, 116.6 (CH), 112.4. MS (ESI) m/z: 248.2 [M + H]+ (calcd for C16H10NO2+ 248.0).O), 1611, 1434 (Ar–), 1113 (C–O). 1H NMR (400 MHz, DMSO) δ: 10.06 (s, 1H), 8.41 (s, 1H), 8.01–7.96 (m, 4H), 7.82 (d, JH–H = 7.5 Hz, 1H), 7.66 (t, JH–H = 7.5 Hz, 1H), 7.46 (d, JH–H = 8.3 Hz, 1H), 7.41 (t, JH–H = 7.5 Hz, 1H). 13C NMR (100 MHz, DMSO) δ: 193.2 (CHO), 159.8, 153.6, 142.5 (CH), 140.9, 136.2, 132.8 (CH), 129.7 (CH), 129.6 (CH), 129.4 (CH), 126.2, 125.2 (CH), 119.7, 116.4 (CH). HR MS (ESI) m/z: 251.0707 [M + H]+ (calcd for C16H11NO3+ 251.0703).O), 1660 (CO), 1605, 1430 (Ar–). 1H NMR (400 MHz, DMSO) δ: 10.08 (s, 1H), 8.17 (s, 1H), 7.93 (s, 1H), 7.76 (dd, JH–H = 7.7 Hz, JH–H = 1.1 Hz, 1H), 7.70 (dd, JH–H = 7.1 Hz, JH–H = 1.9 Hz, 1H), 7.60 (td, JH–H = 7.8 Hz, JH–H = 1.4 Hz, 1H), 7.42–7.33 (m, 4H). 13C NMR (100 MHz, DMSO) δ: 168.9, 160.0, 153.4, 140.9 (CH), 139.6, 135.4, 132.1 (CH), 129.0 (CH), 128.9 (CH), 127.2, 125.0 (CH), 123.6 (CH), 119.8, 119.7 (CH), 119.6 (CH), 116.2 (CH), 24.4 (CH3). HR MS (ESI) m/z: 280.0966 [M + H]+ (calcd for C17H14NO3+ 280.0968).O), 1616, 1577, 1448 (Ar–), 1111 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.74 (s, 1H), 7.70–7.67 (m, 2H), 7.46–7.37 (m, 3H), 7.33–7.31 (m, 2H), 7.24 (dd, JH–H = 7.4 Hz, JH–H = 1.6 Hz, 1H), 2.41 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.8, 151.6, 139.9 (CH), 134.8, 134.1, 132.4 (CH), 128.7 (CH), 128.5 (CH), 128.4 (CH), 128.2, 127.7 (CH), 119.4, 116.1 (CH), 20.8 (CH3). MS (ESI) m/z: 237.2 [M + H]+ (calcd for C16H13O2+ 237.1).O), 1615, 1595, 1570, 1445 (Ar–), 1080 (C–O). 1H NMR (400 MHz, DMSO) δ: 10.66 (s, 1H), 8.15 (s, 1H), 7.69 (d, JH–H = 7.3 Hz, 2H), 7.60 (d, JH–H = 8.5 Hz, 1H), 7.43 (d, JH–H = 7.6 Hz, 2H), 7.37 (t, JH–H = 7.3 Hz, 1H), 6.82 (dd, JH–H = 8.5 Hz, JH–H = 2.2 Hz, 1H), 6.76 (d, JH–H = 2.0 Hz, 1H). 13C NMR (100 MHz, DMSO) δ: 161.7, 160.6, 155.3, 141.6 (CH), 135.6, 130.4 (CH), 128.7 (CH), 128.6 (CH), 128.5 (CH), 122.6, 113.8 (CH), 112.4 (CH), 102.2 (CH). MS (ESI) m/z: 239.1 [M + H]+ (calcd for C15H11O3+ 239.0).O), 1614, 1506, 1464, 1439 (Ar–), 1120 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.75 (s, 1H), 7.68 (d, JH–H = 8.4 Hz, 2H), 7.45–7.41 (m, 3H), 7.39–7.35 (m, 1H), 6.87–6.84 (m, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 162.6, 160.8, 155.3, 139.9 (CH), 135.0, 128.8 (CH), 128.4 (CH), 124.8, 113.3, 112.7 (CH), 100.4 (CH), 55.7 (CH3). MS (ESI) m/z: 253.3 [M + H]+ (calcd for C16H13O3+ 253.1).O), 1604, 1508, 1385, 1361 (Ar–), 1074 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.52 (d, JH–H = 8.8 Hz, 1H), 7.41 (d, JH–H = 7.0 Hz, 2H), 7.35 (d, JH–H = 7.3 Hz, 1H), 7.29–7.25 (m, 2H), 6.85 (dd, JH–H = 8.8 Hz, JH–H = 2.5 Hz, 1H), 6.79 (d, JH–H = 2.2 Hz, 1H), 4.06 (q, JH–H = 7.0 Hz, 2H), 2.23 (s, 3H), 1.43 (t, JH–H = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.7, 161.3, 154.3, 148.0, 134.7, 130.2 (CH), 128.3 (CH), 127.9 (CH), 126.1 (CH), 124.0, 113.9, 112.6 (CH), 101.0 (CH), 64.1 (CH2), 16.5 (CH3), 14.6 (CH3). HR MS (ESI) m/z: 281.1175 [M + H]+ (calcd for C18H17O3+ 281.1172).O), 1614, 1593, 1577, 1446 (Ar–), 1078 (C–O). 1H NMR (400 MHz, DMSO) δ: 10.5 (s, –OH), 7.61 (d, JH–H = 8.8 Hz, 1H), 7.43 (t, JH–H = 7.5 Hz, 2H), 7.36 (t, JH–H = 7.2 Hz, 1H), 7.28 (d, JH–H = 6.9 Hz, 2H), 6.83 (dd, JH–H = 8.8 Hz, JH–H = 2.2 Hz, 1H), 6.74 (d, JH–H = 2.2 Hz, 1H), 2.17 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 161.3, 160.7, 154.3, 148.7, 135.3, 130.7 (CH), 128.4 (CH), 128.0 (CH), 127.5 (CH), 122.7, 113.4 (CH), 112.8, 102.3 (CH), 16.7 (CH3). MS (ESI) m/z: 253.2 [M + H]+ (calcd for C16H13O3+ 253.1).O), 1613, 1445 (Ar–), 1117 (C–O). 1H NMR (400 MHz, DMSO) δ: 7.47 (d, JH–H = 8.7 Hz, 1H), 7.42 (t, JH–H = 7.5 Hz, 2H), 7.35 (t, JH–H = 7.3 Hz, 1H), 7.25 (d, JH–H = 7.0 Hz, 2H), 6.60 (dd, JH–H = 8.7 Hz, JH–H = 2.0 Hz, 1H), 6.46 (d, JH–H = 2.0 Hz, 1H), 6.13 (bs, 2H), 2.14 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 161.1, 154.9, 153.2, 149.1, 135.8, 130.9 (CH), 128.4 (CH), 127.7 (CH), 127.2 (CH), 120.1, 111.8 (CH), 109.5, 98.7 (CH), 16.6 (CH3). MS (ESI) m/z: 252.3 [M + H]+ (calcd for C16H14NO2+ 252.1).O), 1608, 1562, 1448, 1363 (Ar–), 1049 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.60–7.56 (m, 1H), 7.52–7.50 (m, 3H), 7.40 (d, JH–H = 8.3 Hz, 1H), 7.32–7.30 (m, 2H), 7.22 (d, JH–H = 4.1 Hz, 1H), 2.24 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 199.0, 158.4, 153.4, 151.8, 132.7 (CH), 132.5, 129.6 (CH), 128.8 (CH), 128.5 (CH), 128.1 (CH), 127.7, 124.6 (CH), 119.4 (CH), 117.0 (CH), 31.1 (CH3). MS (ESI) m/z: 265.1 [M + H]+ (calcd for C17H13O3+ 265.0).O), 1610, 1452 (Ar–), 1387, 1190 (C–O). 1H NMR (400 MHz, CDCl3) δ: 7.47–7.44 (m, 3H), 7.30 (d, JH–H = 8.6 Hz, 1H), 7.25 (t, JH–H = 7.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 161.4, 152.8, 135.9 (CH), 135.7, 130.4 (CH), 127.2 (CH), 124.1 (CH), 119.5, 116.3 (CH), 28.7 (CH), 21.4 (CH3). MS (ESI) m/z: 189.1 [M + H]+ (calcd for C12H13O2+ 189.0).O), 1591, 1454 (Ar–). 1H NMR (400 MHz, CDCl3) δ: 7.77 (s, 1H), 7.70 (dd, JH–H = 7.0 Hz, JH–H = 1.4 Hz, 2H), 7.59–7.25 (m, 2H), 7.42 (t, JH–H = 7.5 Hz, 2H), 7.35 (t, JH–H = 8.4 Hz, 2H), 7.22 (t, JH–H = 7.3 Hz, 1H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.5, 139.6, 136.8 (CH), 132.4, 130.3 (CH), 128.9 (CH), 128.8 (CH), 128.1 (CH), 128.0 (CH), 122.2 (CH), 120.7, 114.0 (CH), 29.9 (CH3). MS (ESI) m/z: 236.2 [M + H]+ (calcd for C16H14NO+ 236.1).O), 1604, 1598, 1510, 1458 (Ar–), 1246, 1178, 1030. 1H NMR (400 MHz, CDCl3) δ: 7.73 (s, 1H), 7.67 (dd, JH–H = 7.0 Hz, JH–H = 1.9 Hz, 2H), 7.56 (dd, JH–H = 7.8 Hz, JH–H = 1.0 Hz, 1H), 7.51 (td, JH–H = 7.2 Hz, JH–H = 1.4 Hz, 1H), 7.32 (d, JH–H = 8.5 Hz, 1H), 7.21 (td, JH–H = 7.2 Hz, JH–H = 0.6 Hz, 1H), 6.95 (d, JH–H = 8.8 Hz, 2H), 3.83 (s, 3H), 3.76 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.6, 159.5, 139.3, 135.7 (CH), 131.9, 130.2 (CH), 129.9 (CH), 129.2, 128.6 (CH), 122.1 (CH), 120.8, 113.9 (CH), 113.6 (CH), 55.3 (CH3), 29.9 (CH3). MS (ESI) m/z: 266.2 [M + H]+ (calcd for C17H16NO2+ 266.1).O), 1616, 1595, 1508, 1454 (Ar–), 1248, 1211, 1036. 1H NMR (400 MHz, CDCl3) δ: 7.70–7.67 (m, 3H), 7.48 (d, JH–H = 8.6 Hz, 1H), 7.40 (td, JH–H = 7.2 Hz, JH–H = 1.2 Hz, 2H), 7.35–7.31 (m, 1H), 6.81 (dd, JH–H = 8.6 Hz, JH–H = 2.2 Hz, 1H), 6.76 (d, JH–H = 2.2 Hz, 1H), 3.90 (s, 3H), 3.72 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.8, 161.6, 141.2, 137.0, 136.7 (CH), 130.2 (CH), 129.2, 128.8 (CH), 128.1 (CH), 127.7 (CH), 114.9, 109.8 (CH), 98.4 (CH), 55.6 (CH3), 29.9 (CH3). HR MS (ESI) m/z: 266.1179 [M + H]+ (calcd for C17H16NO2+ 266.1176).O), 1579, 1487, 1444, 1415 (Ar–), 1228, 1209, 1118. 1H NMR (400 MHz, CDCl3) δ: 7.73 (d, JH–H = 2.2 Hz, 1H), 7.70–7.67 (m, 3H), 7.63 (dd, JH–H = 8.9 Hz, JH–H = 2.2 Hz, 1H), 7.44 (t, JH–H = 7.6 Hz, 2H), 7.40–7.37 (m, 1H), 7.25 (d, JH–H = 6.7 Hz, 1H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.1, 138.5, 136.3, 135.3 (CH), 133.7, 132.9 (CH), 130.8 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 122.2, 115.7 (CH), 114.9, 30.1 (CH3). MS (ESI) m/z: 314.1 [M + H]+ (calcd for C16H13BrNO+ 314.0).O), 1610, 1464, 1454 (Ar–), 1120, 1028. 1H NMR (400 MHz, DMSO) δ: 11.96 (s, 1H), 8.10 (s, 1H), 7.77–7.72 (m, 3H), 7.50 (td, JH–H = 8.3 Hz, JH–H = 1.2 Hz, 1H), 7.45–7.41 (m, 2H), 7.40 (d, JH–H = 7.3 Hz, 1H), 7.34 (d, JH–H = 8.1 Hz, 1H), 7.20 (td, JH–H = 7.9 Hz, JH–H = 0.8 Hz, 1H). 13C NMR (100 MHz, DMSO) δ: 161.5, 138.8, 138.0 (CH), 136.7, 131.9, 130.6 (CH), 129.1 (CH), 128.6 (CH), 128.4 (CH), 128.3 (CH), 122.3 (CH), 120.0, 115.1 (CH). MS (ESI) m/z: 222.3 [M + H]+ (calcd for C15H12NO+ 222.1).Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 21302042 and 21172055), Department of Henan Province Natural Science and Technology Foundation (No. 142102210410), Natural Science Foundation in Henan Province Department of Education (No. 14B150053), the Program for Innovative Research Team from Zhengzhou (No. 131PCXTD605), and Natural Science Foundation from Technology bureau of Zhengzhou (No. 20130883).Notes and references
- (a) M. A. Musa, J. S. Cooperwood, M. Khan and F. Omar, Curr. Med. Chem., 2008, 15, 2664 CrossRef CAS PubMed; (b) I. Kempen, D. Papapostolou, N. Thierry, L. Pochet, S. Counerotte, B. Masereel, J. M. Foidart, M. J. Reboud-Ravaux, A. Noel and B. Pirotte, Br. J. Cancer, 2003, 88, 1111 CrossRef CAS PubMed; (c) C. Spino, M. Dodier and S. Sotheeswaran, Bioorg. Med. Chem. Lett., 1998, 8, 3475 CrossRef CAS PubMed; (d) P. Anand, B. Singh and N. Singh, Bioorg. Med. Chem., 2012, 20, 1175 CrossRef CAS PubMed; (e) E. Quezada, G. Delogu, C. Picciau, L. Santana, G. Podda, F. Borges, V. Garcia-Moraes, D. Vina and F. Orallo, Molecules, 2010, 15, 270 CrossRef CAS PubMed.
- (a) M. J. Matos, D. Viña, E. Quezada, C. Piciau, G. Delogu, F. Orallo, L. Santana and E. Uriarte, Bioorg. Med. Chem. Lett., 2009, 19, 3268 CrossRef CAS PubMed; (b) M. Matos, D. Viña, C. Picciau, F. Orallo, L. Santana and E. Uriarte, Bioorg. Med. Chem. Lett., 2009, 19, 5053 CrossRef CAS PubMed; (c) M. J. Matos, C. Terán, Y. Pérez-Castillo, E. Uriarte, L. Santana and D. Viña, J. Med. Chem., 2011, 54, 7127 CrossRef CAS PubMed; (d) S. Serra, G. Ferino, M. J. Matos, S. Vázquez-Rodríguez, G. Delogu, D. Viña, E. Cadoni, L. Santana and E. Uriarte, Bioorg. Med. Chem. Lett., 2012, 22, 258 CrossRef CAS PubMed.
- L. M. Kabeya, A. A. Marchi, A. Kanashiro, N. P. Lopes, C. H. T. P. Silva, M. T. Pupo and Y. M. Lucisano-Valim, Bioorg. Med. Chem., 2007, 15, 1515 CrossRef PubMed.
- (a) E. B. B. Ong, N. Watanabe, A. Saito, Y. Futamura, K. H. A. E. Galil, A. Koito, N. Najimudin and H. Osada, J. Biol. Chem., 2011, 286, 14049 CrossRef CAS PubMed; (b) D. Olmedo, R. Sancho, L. M. Bedoya, J. L. Lopez-Perez, E. D. Olmo, E. Munoz, J. Alcami, M. P. Gupta and A. S. Feliciano, Molecules, 2012, 17, 9245 CrossRef CAS PubMed.
- (a) M. de Souza Santos, M. P. F. de Morais Del Lama, L. A. Deliberto, F. da Silva Emery, M. T. Pupo and R. M. Z. G. Naal, Arch. Pharmacal Res., 2013, 36, 731 CrossRef PubMed; (b) H. Zhao, B. Yan, L. B. Peterson and B. S. J. Blagg, ACS Med. Chem. Lett., 2012, 3, 327 CrossRef CAS PubMed.
- (a) C. Wang, C. Wu, J. Zhu, R. H. Miller and Y. Wang, J. Med. Chem., 2011, 54, 2331 CrossRef CAS PubMed; (b) K. G. Reddie, W. H. Humphries, C. P. Bain, C. K. Payne, M. L. Kemp and N. Murthy, Org. Lett., 2012, 14, 680 CrossRef CAS PubMed.
- (a) M. Min and S. Hong, Chem. Commun., 2012, 48, 9613 RSC; (b) M. Khoobi, M. Alipour, S. Zarei, F. Jafarpour and A. Shafiee, Chem. Commun., 2012, 48, 2985 RSC; (c) Y. M. Li, Z. S. Qi, H. F. Wang, X. M. Fu and C. Y. Duan, J. Org. Chem., 2012, 77, 2053 CrossRef CAS PubMed.
- M. J. Matos, S. Vazquez-Rodriguez, F. Borges, L. Santana and E. Uriarte, Tetrahedron Lett., 2011, 52, 1225 CrossRef CAS.
- (a) L. Zhang, T. H. Meng, R. H. Fan and J. Wu, J. Org. Chem., 2007, 72, 7279 CrossRef CAS PubMed; (b) J. B. Meng, M. G. Shen, D. C. Fu, Z. H. Gao, R. J. Wang, H. G. Wang and T. Matsuura, Synthesis, 1990, 719 CrossRef CAS.
- (a) F. Jafarpour, S. Zarei, M. B. A. Olia and N. Jalalimanesh, J. Org. Chem., 2013, 78, 2957 CrossRef CAS PubMed; (b) S. Messaoudi, J. D. Brion and M. Alami, Org. Lett., 2012, 14, 1496 CrossRef CAS PubMed.
- (a) A. Unsinn, S. H. Wunderlich and P. Knochel, Adv. Synth. Catal., 2013, 355, 989 CrossRef CAS; (b) M. Mosrin, G. Monzon, T. Bresser and P. Knochel, Chem. Commun., 2009, 37, 5615 RSC.
- (a) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 16, 11212 Search PubMed.
- F. Jafarpour, M. B. a. Olia and H. Hazrati, Adv. Synth. Catal., 2013, 355, 3407 CrossRef CAS.
- P. Chauhan, M. Ravi, S. Singh, P. Prajapati and P. P. Yadav, RSC Adv., 2016, 6, 109 RSC.
- Z. J. She, Y. Shi, Y. M. Huang, Y. Y. Cheng, F. J. Song and J. S. You, Chem. Commun., 2014, 50, 13914 RSC.
- F. Jafarpour, H. Hazrati, N. Mohasselyazdi, M. Khoobi and A. Shafiee, Chem. Commun., 2013, 49, 10935 RSC.
- S. Martins, P. S. Branco, M. C. de la Torre, M. A. Sierra and A. Pereira, Synlett, 2010, 19, 2918 Search PubMed.
- (a) J. M. Liu, X. Zhang, L. J. Shi, M. W. Liu, Y. Y. Yue, F. W. Li and K. L. Zhao, Chem. Commun., 2014, 50, 9887 RSC; (b) K. V. Sashidhara, G. R. Palnati, S. R. Avula and A. Kumar, Synlett, 2012, 4, 611 CrossRef; (c) H. Y. Zeng and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 13862 CrossRef CAS PubMed.
- (a) J. W. Yuan, Q. Y. Yin, L. R. Yang, W. P. Mai, P. Mao, Y. M. Xiao and L. B. Qu, RSC Adv., 2015, 5, 88258 RSC; (b) A. Nakatani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 1377 CrossRef CAS PubMed; (c) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed.
- (a) A. S. Demir, O. Reis and M. Emrullahoglu, J. Org. Chem., 2003, 68, 578 CrossRef CAS PubMed; (b) A. Dickschat and A. Studer, Org. Lett., 2010, 12, 3972 CrossRef CAS PubMed; (c) G. B. Yan, M. H. Yang and X. M. Wu, Org. Biomol. Chem., 2013, 11, 7999 RSC.
- (a) A. S. Demir and H. Findik, Tetrahedron, 2008, 64, 6196 CrossRef CAS; (b) A. S. Demir, H. Findik, N. Saygili and N. T. Subasi, Tetrahedron, 2010, 66, 1308 CrossRef CAS.
- S. Paul, K. Pradhan, S. Ghosh, S. K. De and A. R. Das, Adv. Synth. Catal., 2014, 356, 1301 CrossRef CAS.
- Y. S. Jiang, W. Z. Chen and W. M. Lu, Tetrahedron, 2013, 69, 3669 CrossRef CAS.
- O. V. Skripskaya, N. O. Feilo, A. O. Neshchadin, O. V. Elenich, R. Z. Lytvyn, N. D. Obushak and P. I. Yagodinets, Russ. J. Org. Chem., 2013, 49, 1655 CrossRef CAS.
- S. S. AI-Shihry, Molecules, 2004, 9, 658 CrossRef.
- P. Chauhan, M. Ravi, S. Singh, P. Prajapati and P. P. Yadav, RSC Adv., 2016, 6, 109 RSC.
- S. Kim, D. J. Kang, C. H. Lee and P. H. Lee, J. Org. Chem., 2012, 77, 6530 CrossRef CAS PubMed.
- N. M. Naik and K. R. Desai, J. Inst. Chem., 1988, 60, 179 CAS.
- H. Y. Sun, Y. H. Zhang, F. F. Guo, Y. Z. Yan, C. F. Wan, Z. G. Zha and Z. Y. Wang, Eur. J. Org. Chem., 2012, 3, 480 CrossRef.
- N. Toshihira, H. Etsuro and O. Nobuko, Yakugaku Zasshi, 1954, 74, 250 Search PubMed.
- L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. F. Du and K. Zhao, Org. Lett., 2013, 15, 2906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Part of the experimental detail, and NMR spectra data. CCDC 1447043. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra04787d |
| This journal is © The Royal Society of Chemistry 2016 |