DOI:
10.1039/C6RA04762A
(Paper)
RSC Adv., 2016,
6, 38957-38963
Hydrophobic association hydrogels based on N-acryloyl-alanine and stearyl acrylate using gelatin as emulsifier†
Received
23rd February 2016
, Accepted 12th April 2016
First published on 13th April 2016
Abstract
Poly(N-acryloyl-alanine)-based hydrophobic association hydrogels were prepared through free radical polymerization of N-acryloyl-alanine (NAA) with ammonium persulfate as initiator, in which hydrophobic monomer stearyl acrylate underwent simultaneous micellar polymerization in the presence of gelatin as emulsifier. Fourier transform infrared spectra and scanning electron microscopy demonstrated the formation and microscopic structure of the resulting hydrogels. The hydrophobic association enabled the hydrogels to exhibit desirable toughness and could be molded into diverse shapes without breaking. Uniaxial tensile test and cyclic tensile test also demonstrated the high toughness of the hydrogels. The chiral monomer (NAA)-derived polymer chains rendered the hydrogels with optical activity, according to circular dichroism spectra. More fascinatingly, the hydrogels demonstrated shape memory behavior due to the hydrophobic poly(stearyl acrylate) domains. Therefore the tough chiral hydrogels are expected to find significant applications in tissue engineering and other biomedical fields.
1. Introduction
Hydrogels have received growing attention due to their biodegradability, intelligence, and ability to absorb and retain a large amount of water.1–3 These features make hydrogels quite similar to human tissues4,5 and have been investigated as biomaterials (e.g. for bio-adhesives,6 tissue engineering,7,8 cartilage repair,9 drug delivery,10 embolic agents,11 and injectable hydrogels12,13). However, generally hydrogels demonstrate poor mechanical strength and lack stretchability, which are critical for practical applications.14,15 For example, the hydrogels need to afford enough stress without being damaged when used as tissue engineering scaffolds. They should withstand long-term repeated use when utilized as absorbents. Therefore, in recent years a number of techniques have been proposed for toughening hydrogels,16,17 by which a variety of hydrogels have been reported, e.g. double network hydrogels,18 topological hydrogels,19 nanocomposite hydrogels,20,21 and macromolecular microsphere composite hydrogels.22 The essential idea in these methods is to dissipate the energy and thereby to improve the strength and toughness of hydrogels.23
A facile way to dissipate the energy is to build reversible crosslinks instead of permanent crosslinks inside hydrogel networks. Upon application of external stress, reversible crosslinks in a hydrogel can move inside its networks and balance polymer chains to dissipate the stress.24,25 Hydrophobic interactions have been proved to be one of the effective ways to toughen hydrogels.26,27 Reportedly, hydrophobically modified hydrogels were prepared by copolymerization of hydrophilic monomer with a relatively small amount of hydrophobic comonomer with the aid of emulsifier via the so-called free radical micellar polymerization.28,29 Micellar polymerizations are favorable for preparing hydrogels whose properties can be easily tuned by controlling the hydrophobic components formed inside micelles. The resulting hydrogels are of significant promise because their properties could be readily adjusted just by changing the ratio of hydrophobic and hydrophilic monomers. Jiang et al.30,31 synthesized tough hydrogels by the copolymerization of hydrophilic acrylamide and a small amount of hydrophobic octylphenol polyoxyethylene acrylate with sodium dodecyl sulfate (SDS) as emulsifier. The as-obtained hydrogels possessed remarkable mechanical strength. Okay et al.32–34 prepared a series of tough hydrogels based on hydrophobic interaction which even provided some fascinating properties e.g. self-healing and shape memory properties. Herein, we hope to point out that in the above reported hydrogels, small molecular emulsifier, e.g. SDS was used, and it might be toxic and thus produce some unfavorable influence on practical applications.27 To circumvent this problem, biomacromolecules may be a good choice instead of SDS.35 Among the widely used biomacromolecules, gelatin is ampholytic and has been extensively used in pharmaceutical and food industries. Especially, gelatin macromolecule chains could stabilize the oil phase in water and thus could be used as emulsifier.36,37
In the present study, we prepared tough, chiral hydrogels for the first time through simultaneous free radical polymerization of chiral hydrophilic monomer N-acryloyl-alanine (NAA) and free radical micellar polymerization of hydrophobic monomer stearyl acrylate (C18) in aqueous media at 60 °C by using gelatin as emulsifier and ammonium persulfate as initiator. Gelatin was used as emulsifier for performing the micellar polymerization of C18 and it is also expected to improve the biocompatibility of the hydrogels. The as-obtained hydrophobic association hydrogels (HAGs) demonstrated toughness due to the hydrophobic interactions among stearyl acrylate-derived polymer chains. The chiral monomer NAA derived from L-alanine and acryloyl chloride endowed the hydrogels with intriguing optical activity. Herein, it is important to point out that the major driving force for us to use NAA as starting material lies in that amino acid-derived polymers (e.g. alanine-based polymers38–40) have constituted a unique class of polymers currently drawing increasing attention. The study along this direction may provide novel chiral polymer materials as demonstrated by our earlier studies.41,42
2. Experimental section
2.1 Materials
L-Alanine and acryloyl chloride were purchased from Aladdin (China). Gelatin (type A, from porcine skin with a gel strength about 300 g Bloom) and stearyl acrylate (C18) were obtained from Sigma-Aldrich Company. Ethyl acetate, anhydrous ethanol, sodium hydroxide (NaOH), hydrochloric acid (HCl), and ammonium persulfate (APS) were purchased from Beijing Chemical Reagents Company. All the reagents obtained were of analytic grade and used without further purification.
2.2 Synthesis of chiral monomer NAA
The chiral monomer NAA was synthesized according to our earlier investigations.43 The major procedure is stated as below. 2.784 g L-alanine was added in 12.5 mL 0.2 mol L−1 NaOH aqueous solution at 25 °C; and then 2.5 mL acryloyl chloride was dropwise added in the reaction system in an ice-bath after bubbled with N2 for 20 min. The white precipitate appeared by adding 10 mL 2 mol L−1 HCl aqueous solution after reacting for 2 h in ice-bath and then for another 1 h at room temperature. After extracting five times with ethyl acetate and evaporation of the solvent, we could obtain the coarse product. After further purification by recrystallization from ethanol at 0 °C and dried under 50 °C vacuum oven, pure L-NAA monomer was obtained.
2.3 Preparation of hydrophobic association hydrogels
Hydrophobic association hydrogels (HAGs) were synthesized by free radical copolymerization of hydrophilic NAA with hydrophobic C18 at 60 °C for 24 h by using macromolecular gelatin as emulsifier and APS as initiator. Herein, the comonomers' total concentration was fixed at 20% w/v and the content of the hydrophobic monomer in the total monomers was 2%, 3% and 4% (in mol), and the corresponding hydrogels were denoted as HAG2%, HAG3% and HAG4%, respectively. The detailed preparation of the hydrogels is stated below by taking HAG2% as example. Gelatin (0.75 g) was dissolved in 5.3 mL deionized water at 40 °C and stirred for 2 h to obtain a transparent solution and then hydrophobic monomer C18 (0.204 mmol) was added in the solution. After C18 was uniformly emulsified by gelatin under stirring, NAA solution (9.996 mmol NAA in 2 mL NaOH solution) was added in the mixture. Then initiator APS was added and the reaction emulsion was bubbled with N2 for 10 min. The mixture solution was transferred quickly to a plastic tube (6 mm diameter for tensile test and 10 mm diameter for manual compression) and sealed immediately. The sealed plastic tube was immediately placed in a 60 °C water bath and then polymerization began and lasted for 24 h. Hydrogels with excellent mechanical property were thus obtained. For comparison, hydrogel samples without gelatin and C18 were also prepared under comparable conditions (see Table 1).
Table 1 Parameters for preparing hydrophobic association hydrogels (HAG)a
| HAG |
C18 ratio (mol%) |
Gelatin (g) |
C18 (mmol) |
NAA (mmol) |
APS (mmol) |
| HAG2%, HAG3%, and HAG4% showed satisfactory toughness; blank-1: only sticky product rather than hydrogel formed; blank-2: the hydrogel was too weak and could not be stretched. |
| HAG2% |
2% |
0.75 |
0.204 |
9.996 |
0.204 |
| HAG3% |
3% |
0.75 |
0.303 |
9.797 |
0.202 |
| HAG4% |
4% |
0.75 |
0.399 |
9.576 |
0.199 |
| Blank-1 |
3% |
0 |
0.303 |
9.797 |
0.202 |
| Blank-2 |
0 |
0.75 |
0.303 |
9.797 |
0.202 |
2.4 Characterization of the emulsions
The emulsions (C18 in gelatin aqueous solution at 40 °C) were observed by optical microscopy equipped with a Canon digital camera to get photos of the droplets.44 Specifically, a drop of emulsion was placed on a piece of glass slide and then covered with another piece of cover glass. After setting the magnification as 400 and regulating the knob to find the clear photograph, the morphology could be observed by the digital camera. To further demonstrate the emulsification property of gelatin, the solution only containing C18 and water was also viewed by the optical microscopy.
2.5 Characterization of the hydrogels
Fourier transform infrared spectra (FT-IR) were recorded on a TENSOR 27 spectrophotometer (KBr tablet for powder specimen and ATR for dried hydrogels). Circular dichroism (CD) and UV-vis absorption spectra were measured on JASCO J-810 spectropolarimeter at room temperature, by placing a certain amount of water-swollen hydrogels between two pieces of quartz glass. The structure of hydrogels was observed by scanning electron microscopy (SEM, Hitachi S-4800). For this purpose, the swollen hydrogels were frozen in liquid nitrogen and then freeze-dried in the freeze direr for 48 h before observation.
2.6 Tensile tests
Uniaxial tensile tests were performed on electric tensile tester (SANS, China) with a 500 N load cell at room temperature.45 The samples were cylindrical hydrogels of about 6 mm in diameter and 25 mm in length, and the sample length between the clamps was 14 mm and the tensile speed was 500 mm min−1. Cyclic tests were also conducted, and the maximum elongation was fixed at 500% using HAG4% as example. When the hydrogel was stretched to 500% elongation, the sample was unloaded until the stress became zero. The second and third loading–unloading cycles were conducted immediately without any stay. Herein, the corresponding three loading–unloading curves were noted as 41, 42 and 43. Meanwhile, after a waiting time for 15 min, the same HAG4% sample was performed for the fourth loading–unloading cycle, and we named the recorded stress–strain curve as 44.
2.7 Shape memory behavior
The cylindrical hydrogel sample (HAG4%) with a diameter of 6 mm and length of 6 cm was used to examine the shape memory behavior.34,46 The hydrogel was wrapped with plastic wrap and immersed in 60 °C water for 3 h (the reason for the usage of plastic wrap was to guarantee the hydrogel contacting but not absorbing hot water), which facilitated the shape changing of the hydrogel. Then the hydrogel was twisted on a glass rod to form spiral shape, and this temporary spiral shape was fixed by cooling the sample to ambient temperature and further equilibrating overnight. After that the deformed hydrogel sample was immersed in a water bath at 60 °C for recovering to the original shape. To measure the shape recovery ratio R, the sample with a original length of 2 cm was strentched to 7 cm, and then the sample length L was recorded at 1 min intervals. R was calculated by the equation: R = (7 − L)/(7 − 2) × 100%.
3. Results and discussion
3.1 Strategy to prepare the hydrogels
In the present study, we designed and successfully prepared tough chiral hydrogels via free radical (micellar) polymerization approach. The aims are to improve the hydrogels' mechanical properties by hydrophobic interaction and to improve the biocompatibility by introducing biopolymer gelatin as macromolecular emulsifier. The materials used for preparing the hydrogels include only emulsifier gelatin, hydrophilic monomer NAA, hydrophobic monomer C18, and initiator APS. As we know, hydrogels were three-dimensional networks crosslinked by chemical and/or physical interactions. Due to the emulsifier property of gelatin mentioned above and the absence of any crosslinker, the mechanism for forming the hydrogels is considered as free radical micellar polymerization.28,29 The preparative strategy is illustratively presented in Scheme 1, where gelatin acted as emulsifier for the hydrophobic monomer. Herein, we highlight that the hydrophobic aggregation interaction served as crosslinking points for forming the hydrogel networks.
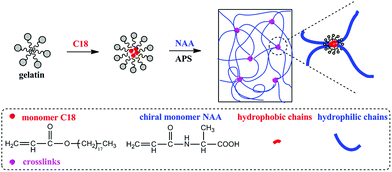 |
| | Scheme 1 Schematic illustration for forming tough hydrogels by hydrophobic association effect. | |
In the strategy presented in Scheme 1, the key factor for forming hydrophobic association hydrogels is the presence of hydrophobic polymer domains derived from C18 in aqueous solution, with the aid of emulsifying function of gelatin. To elucidate this hypothesis, optical microscope was used to observe the emulsion comprising C18 and gelatin, and the corresponding photographs are shown in Fig. 1b–d. Fig. 1a shows the C18 droplets in the blank test only containing the hydrophobic monomer C18 and water (without any gelatin). In this case, the hydrophobic monomer C18 assumed a “sheet” form and floated on water, indicating that C18 could not be dispersed in water. However, C18 in the gelatin aqueous solution (Fig. 1b–d) became spherical and well dispersed in the aqueous system. This is due to the emulsification effect of gelatin. Therefore C18 could be dispersed in the gelatin aqueous solution and stabilized by gelatin macromolecules.
 |
| | Fig. 1 Optical micrographs of the emulsion droplets (a) only C18 and water (no gelatin); (b–d) C18 in the gelatin aqueous solution with C18 content: 2, 3 and 4 mol%, respectively. | |
Three hydrophobic association hydrogels were produced by changing the hydrophobic/hydrophilic monomer ratio, and the corresponding parameters and hydrogels as-formed are summarized in Table 1. The hydrogels formed by the physical hydrophobic interactions could be molded into arbitrary shapes due to the reversible physical crosslinking points (Fig. 2). For example, the hydrogels (taking HAG3% as a representative) could be molded as a circle, a U form, and a cross without any damage (Fig. 2a). Furthermore, the swelling ability of the three hydrogels was measured. The swelling ratio of them was approximately 150 (HAG2%), 130 (HAG3%), and 120 times (HAG4%). (The swelling ratio was determined with the corresponding dried hydrogel as control sample). Fig. S1 (ESI†) presents a typical photograph for HAG2% after swelling in water.
 |
| | Fig. 2 Digital photographs of HAG3% with excellent mechanical toughness. The hydrogel exhibited high level of deformation: (a) molded into arbitrary shapes, (b) stretched by hand, (c) compressed manually to a certain degree without any damage. | |
To further testify the free radical micellar copolymerization and the hydrophobic association effect, two blank experiments respectively named as blank-1 and blank-2 were also conducted (Table 1). In blank-1, all the other conditions for preparing HAG3% kept unchanged but without adding gelatin; in blank-2, all the other conditions for preparing HAG3% kept unchanged but without adding C18 (the products from the two blank tests are also named as blank-1 and blank-2, respectively). The photographs for the products (blank-1 and blank-2) after polymerization at 60 °C for 24 h are shown in Fig. S2 (ESI†). For blank-1, hydrophobic C18 could not be dispersed in aqueous solution; and only a sticky material rather than hydrogel was formed after polymerization, due to the lacking of gelatin. As for blank-2, even though hydrogel could be formed, however the hydrogel was too weak to be stretched due to the lack of C18 which otherwise should be responsible for forming physical crosslinks. The products in blank-1 and blank-2 clearly demonstrate the roles played by gelatin and C18 in forming the designed hydrogels. The C18-derived components inside the hydrogels provide hydrophobic interactions due to their insolubility in water and accordingly provide the desired strength for the hydrogels. In addition, we could compress the hydrogels with pressure by two fingers nearly to 50% of its original height without any visible cracks or breaks along the hydrogel (Fig. 2c1–c3). All these properties testified the toughness (flexible and elastic) of the obtained chiral hydrogels. The tensile properties will be quantitatively discussed later on.
3.2 Characterization of the hydrogels
To preliminarily confirm the formation of hydrophobic association hydrogels, HAG2% was taken as a representative of the hydrogels, together with the reactants, were measured by FT-IR spectroscopy. The FT-IR spectra are shown in Fig. 3. A comparison among the spectra of gelatin (spectrum a), stearyl acrylate (C18, spectrum b), NAA (spectrum c), and the product HAG2% (spectrum d) provides us a strong evidence for the successful formation of the hydrogels. Specifically, the peaks at 1611 cm−1 (Fig. 3b) and 1602 cm−1 (Fig. 3c), which are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in hydrophobic monomer C18 and hydrophilic monomer NAA, disappeared completely in the hydrogel network (Fig. 3d), indicating that the double bonds were consumed and polymerization took place in the monomers. Meanwhile, the corresponding characteristic peaks in NAA and C18 could also be seen in the hydrogel network. The appearance of the characteristic peaks at 1732 (CO absorption), 1651 and 1550 cm−1 (amide group) of NAA and the peaks at 2925 and 2854 cm−1 (C–H stretching) of C18 demonstrated the presence of the monomer units in the hydrogel. All the observations discussed above further confirmed the formation of hydrogels.
C double bonds in hydrophobic monomer C18 and hydrophilic monomer NAA, disappeared completely in the hydrogel network (Fig. 3d), indicating that the double bonds were consumed and polymerization took place in the monomers. Meanwhile, the corresponding characteristic peaks in NAA and C18 could also be seen in the hydrogel network. The appearance of the characteristic peaks at 1732 (CO absorption), 1651 and 1550 cm−1 (amide group) of NAA and the peaks at 2925 and 2854 cm−1 (C–H stretching) of C18 demonstrated the presence of the monomer units in the hydrogel. All the observations discussed above further confirmed the formation of hydrogels.
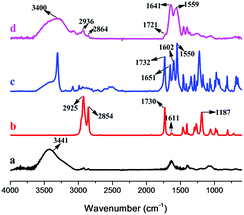 |
| | Fig. 3 Typical FT-IR spectra of (a) gelatin, (b) C18, (c) NAA and (d) HAG 2% (as representative for the HAGs). | |
The hydrogels were immersed in water to reach water-uptake equilibrium, and then frozen-dried in the freeze direr in order to observe the structure of the hydrogels by SEM. The relevant SEM images are illustrated in Fig. 4. The pore structures of the hydrogels were remained after freeze-drying water. The pores tended to become smaller with the increase in the hydrophobic monomer ratio, which increased the crosslink density of the hydrogels.
 |
| | Fig. 4 SEM images of the cross-section of hydrophobic association hydrogels. (a) HAG2%; (b) HAG3%; (c) HAG4%. | |
3.3 Optical activity of the hydrogels
The present study aimed to prepare tough chiral hydrogels through free radical micellar polymerization. Herein, the hydrophilic monomer NAA, synthesized by the reaction between chiral L-alanine and acryloyl chloride, in theory should endow the hydrogels with the desired optical activity. Hence, the water-swollen equilibrium hydrogels were placed between two pieces of quartz glass and subjected to circular dichroism (CD) and UV-vis absorption spectroscopy measurement at room temperature, since CD and UV-vis spectroscopies are effective to characterize the optical activity of materials.47–50 The hydrogels showed considerable CD signals around 225 nm in Fig. 5a, indicating the optical activity of the hydrogels. The corresponding UV-vis spectroscopy presented in Fig. 5b also had absorptions at wavelength below 250 nm. To acquire deep understanding of the chiral hydrogels, we also measured the CD and UV-vis spectra of gelatin. The related spectra are also illustrated in Fig. 5. It shows the CD spectra of the hydrogels are quite different from that of gelatin. Referring to our earlier studies concerning chiral hydrogels constructed by NAA,41 we conclude that the optical activity of the hydrogels was originated majorly in the chiral monomer NAA.
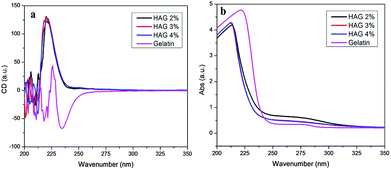 |
| | Fig. 5 CD (a) and UV-vis (b) spectra of the hydrogels swelled in deionized water at ambient temperature. | |
3.4 Mechanical properties of the hydrogels
The mechanical properties of the obtained hydrogels were investigated by tensile test. Fig. 6a demonstrates the specific photographs of the hydrogel HAG2% before the tensile test and the just before rupture in the tensile test using electric tensile tester. The photographs vividly manifested the mechanical properties of the hydrophobic association hydrogels. Fig. 6b shows the corresponding stress–strain curves of the three hydrogels (HAG2%, HAG3%, and HAG4%, see Table 1). As the content of C18 increased (in the order of HAG2%, HAG3%, and HAG4%), the corresponding stress and strain increased and reached the maximum values of 76 kPa and 750%, respectively (HAG4%). When subjected to external load, the hydrophobic association crosslinks inside the hydrogels slided and thus dissipated the mechanical energy inside the hydrogel networks.13 Therefore, the hydrophobic association hydrogels showed remarkable toughness, similar to the properties of elastomers, which is highly desirable from the view point of practical applications.
 |
| | Fig. 6 Typical photographs of HAG2% before the tensile test (a1) and just before rupture (a2); (b) stress–strain curves of the hydrogels. In (a1) and (a2), the hydrogel sample was highlighted by red line box. | |
Cyclic tensile tests at a certain strain were also performed on HAG4% for further understanding the hydrogels, and the obtained loading/unloading curves are shown in Fig. 7. The results show that the hydrogel exhibited evident stress softening phenomenon, similar to elatomer materials, when the loading/unloading tensile test was performed ongoing without any interval time (see curves 41, 42, and 43). Interestingly, after finishing the third cyclic test and remaining a 15 min waiting time, the obtained 44 curve (the fourth test) in Fig. 7 was close to the first curve 41 and the relevant values were also higher than the last two curves (42 and 43). During the first three tests, the hydrogel was continuously tested without any pause, so the hydrogel did not have time to recover to its original state. When the hydrogel was left for a 15 min waiting time, the hydrogel network, at least partially, recovered to its original network (curve 44). Thus the cyclic tests confirm the energy dissipation in the hydrogels and further the presence of reversible hydrophobic crosslinks in the hydrogel networks.51 This consideration is further manifested by the performance of shape memory of the hydrogels, as to be reported next.
 |
| | Fig. 7 Four successive loading/unloading cycles of hydrogel HAG4% with a fixed strain 500%. The first three curves (41, 42, and 43) were conducted immediately after the preceding tensile test. The fourth curve (44) was conducted after 15 min of waiting time. | |
3.5 Shape memory performance of the hydrogels
The presence of hydrophobic interactions may provide the hydrogels with some other interesting properties, such as self-healing and shape memory behavior.31,34,46 According to the earlier reports in literature, the hydrogels formed by stearyl acrylate bearing long alkyl chains could be able to crystallize upon self-assembly into hydrophobic domains. These crystalline domains were very stable and could be disrupted only upon heating above the melting temperature.52 The melting temperature (Tm) of the hydrophobic C18 polymer is about 50 °C,53–55 and therefore the corresponding thermal-induced shape memory behavior of HAG4% was conducted at 60 °C. The results are depicted in Fig. 8.
 |
| | Fig. 8 Photographs demonstrating thermal-induced shape memory process of HAG4% from the temporary spiral shape to the permanent rod shape. | |
In Fig. 8, the permanent rod shape hydrogel was treated in 60 °C water and thus could be easily deformed into the temporary spiral shape by being wrapped on a glass rod. For the hydrogels, the crystalline domains formed by the long alkyl groups in C18 facilitated to fix the temporary shape at room temperature.34,56 The above temporary spiral shape was fixed at ambient temperature within 12 h. After putting it in hot water (60 °C) again, the temporary spiral shape gradually recovered to nearly its original rod shape within 180 s. We further quantitatively measured the recovery ratio R (as defined above) of the three hydrogels and the results are presented in Fig. 9. It shows the shape recovery ratio (R) of the hydrogels at different time. All the three hydrogels showed a high shape recovery ratio (>80%). When the temperature was above the melting temperature of the crystalline domains formed by C18-derived polymer chains, the crystalline domains melted. Accordingly, the physically crosslinked network structures resulting from the hydrophobic association enabled the hydrogels to recover nearly to the original length.
 |
| | Fig. 9 Curves of shape recovery ratio (R) vs. time of the hydrogels. The detailed conditions are presented in Experimental section. | |
4. Conclusion
Tough chiral hydrophobic association hydrogels were prepared through free radical micellar polymerization of stearyl acrylate with gelatin as emulsifier, together with the radical polymerization of NAA in the same aqueous polymerization system. Herein, the hydrophobic association domains acted as physical crosslinking points, by which to form tough hydrogels. The hydrogels could be strentched and compressed, and also notably, the physically crosslinked network structures enabled the hydrogels to exhibit interesting shape memory behavior. The chiral monomer NAA endowed the hydrogels with optical activity, according to CD spectra. All these remarkable advantages facilitated the present hydrogels to be further investigated for establishing advanced functional biomaterials. Especially worthy to note is the presence of gelatin in the hydrogels, which is expected to afford the hydrogels with desirable biocompatibility. The potential uses of the chiral hydrogels in chiral-related areas also deserve much more attention. Our research is currently undergoing along the directions.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21474007, 21274008, 21174010), the Funds for Creative Research Groups of China (51221002), and the “Specialized Research Fund for the Doctoral Program of Higher Education” (SRFDP 20120010130002).
Notes and references
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- T. Jungst, W. Smolan, K. Schacht, T. Scheibel and J. Groll, Chem. Rev., 2016, 116, 1496 CrossRef CAS PubMed.
- K. Yue, G. T. de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254 CrossRef CAS PubMed.
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. Van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020 CrossRef CAS PubMed.
- J. C. Cuggino, C. B. Contreras, A. J. Kairuz, B. A. Maletto and C. I. A. Igarzabal, Mol. Pharm., 2014, 11, 2239 CrossRef CAS PubMed.
- C. Ghobril and M. W. Grinstaff, Chem. Soc. Rev., 2015, 44, 1820 RSC.
- J.-F. Xing, M.-L. Zheng and X.-M. Duan, Chem. Soc. Rev., 2015, 44, 5031 RSC.
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717 CrossRef CAS PubMed.
- W. Li, D. Wang, W. Yang and Y. Song, RSC Adv., 2016, 6, 20166 RSC.
- E. P. da Silva, M. R. Guilherme, F. P. Garcia, C. V. Nakamura, L. Cardozo-Filho, C. G. Alonso, A. F. Rubira and M. H. Kunita, RSC Adv., 2016, 6, 19060 RSC.
- X. Su, L. Bu, H. Dong, S. Fu, R. Zhuo and Z. Zhong, RSC Adv., 2016, 6, 2904 RSC.
- D. Maitland, S. B. Campbell, J. Chen and T. Hoare, RSC Adv., 2016, 6, 15770 RSC.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973 CrossRef CAS.
- J. Y. Sun, X. H. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- K. R. Shull, Nature, 2012, 489, 36 CrossRef CAS PubMed.
- X. J. Liu, H. Q. Li, B. Y. Zhang, Y. J. Wang, X. Y. Ren, S. Guan and G. H. Gao, RSC Adv., 2016, 6, 4850 RSC.
- Y. Lin, D. He, Z. Chen, L. Wang and G. Li, RSC Adv., 2016, 6, 12479 RSC.
- T. C. Suekama, J. Hu, T. Kurokawa, J. P. Gong and S. H. Gehrke, ACS Macro Lett., 2013, 2, 137 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485 CrossRef CAS.
- M. X. Shen, L. Li, Y. M. Sun, J. Xu, X. H. Guo and R. K. Prud’homme, Langmuir, 2014, 30, 1636 CrossRef CAS PubMed.
- Y. Piao and B. Chen, RSC Adv., 2016, 6, 6171 RSC.
- F. Z. Jiang, T. Huang, C. C. He, H. R. Brown and H. L. Wang, J. Phys. Chem. B, 2013, 117, 13679 CrossRef CAS PubMed.
- M. Shibayama, Soft Matter, 2012, 8, 8030 RSC.
- J. Q. Xu, D. A. Bohnsack, M. E. Mackay and K. L. Wooley, J. Am. Chem. Soc., 2007, 129, 506 CrossRef CAS PubMed.
- Y. Wei, Y. Wang, C. Wei, Q. Zhao, Y. Yan, J. Yang and J. Huang, RSC Adv., 2015, 5, 106005 RSC.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449 CrossRef CAS.
- W. B. Li, H. Y. An, Y. Tan, C. G. Lu, C. Liu, P. C. Li, K. Xu and P. X. Wang, Soft Matter, 2012, 8, 5078 RSC.
- E. J. Regalado, J. Selb and F. Candau, Macromolecules, 1999, 32, 8580 CrossRef CAS.
- P. Kujawa, A. A. Hayet, J. Selb and F. Candau, Macromolecules, 2006, 39, 384 CrossRef CAS.
- G. Q. Jiang, C. Liu, X. L. Liu, G. H. Zhang, M. Yang and F. Q. Liu, Macromol. Mater. Eng., 2009, 294, 815 CrossRef CAS.
- G. Q. Jiang, C. Liu, X. L. Liu, Q. R. Chen, G. H. Zhang, M. Yang and F. Q. Liu, Polymer, 2010, 51, 1507 CrossRef CAS.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997 CrossRef CAS.
- U. Gulyuz and O. Okay, Soft Matter, 2013, 9, 10287 RSC.
- C. Bilici and O. Okay, Macromolecules, 2013, 46, 3125 CrossRef CAS.
- X. Zhang, Y. H. Yang, J. R. Yao, Z. Z. Shao and X. Chen, ACS Sustainable Chem. Eng., 2014, 2, 1318 CrossRef CAS.
- J. Olijve, F. Mori and Y. Toda, J. Colloid Interface Sci., 2001, 243, 476 CrossRef CAS.
- L. Lobo, J. Colloid Interface Sci., 2002, 254, 165 CAS.
- K. Bauri, K. D. Sayala, R. S. Roy and P. De, Eur. Polym. J., 2015, 73, 237 CrossRef CAS.
- K. Bauri, A. Narayanan, U. Haldar and P. De, Polym. Chem., 2015, 6, 6152 RSC.
- Y. Shen, G. Li, Y. Ma, D. Yu, J. Sun and Z. Li, Soft Matter, 2015, 11, 7502 RSC.
- P. Xie, X. Liu, R. Cheng, Y. P. Wu and J. P. Deng, Ind. Eng. Chem. Res., 2014, 53, 8069 CrossRef CAS.
- R. Cheng, J. Liu, P. Xie, Y. P. Wu and J. P. Deng, Polymer, 2015, 68, 246 CrossRef.
- L. Shi, P. Xie, Z. M. Li, Y. P. Wu and J. P. Deng, Macromol. Chem. Phys., 2013, 214, 1375 CrossRef CAS.
- J. Surh, E. A. Decker and D. J. McClements, Food Hydrocolloids, 2006, 20, 596 CrossRef CAS.
- A. K. Gaharwar, S. A. Dammu, J. M. Canter, C. J. Wu and G. Schmidt, Biomacromolecules, 2011, 12, 1641 CrossRef CAS PubMed.
- J. K. Hao and R. A. Weiss, ACS Macro Lett., 2013, 2, 86 CrossRef CAS.
- K. Maeda, M. Muto, T. Sato and E. Yashima, Macromolecules, 2011, 44, 8343 CrossRef CAS.
- W. F. Li, X. Liu, G. Y. Qian and J. P. Deng, Chem. Mater., 2014, 26, 1948 CrossRef CAS.
- F. Yao, D. Y. Zhang, C. H. Zhang, W. T. Yang and J. P. Deng, Bioresour. Technol., 2013, 129, 58 CrossRef CAS PubMed.
- C. Song, C. H. Zhang, F. J. Wang, W. T. Yang and J. P. Deng, Polym. Chem., 2013, 4, 645 RSC.
- X. H. Zhao, Soft Matter, 2014, 10, 672 RSC.
- O. E. Philippova, A. S. Andreeva, A. R. Khokhlov, A. Islamov, A. I. Kuklin and V. I. Gordeliy, Langmuir, 2003, 19, 7240 CrossRef CAS.
- A. Matsuda, J. Sato, H. Yasunaga and Y. Osada, Macromolecules, 1994, 27, 7695 CrossRef CAS.
- F. Dutertre, P.-Y. Pennarun, O. Colombani and E. Nicol, Eur. Polym. J., 2011, 47, 343 CrossRef CAS.
- Z. Ren, W. Hu, C. Liu, S. Li, X. Niu and Q. Pei, Macromolecules, 2016, 49, 134 CrossRef CAS.
- K. Inomata, T. Terahama, R. Sekoguchi, T. Ito, H. Sugimoto and E. Nakanishi, Polymer, 2012, 53, 3281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04762a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 









