
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of non-ionic AB-diblock nanoparticles prepared by RAFT dispersion polymerization with polymerization-induced self-assembly†
Yiwen
Pei
ab,
Kevin
Jarrett
c,
Leonardo Gutierrez
Garces
bde,
Martin
Saunders
f,
Jean-Philippe
Croue
bd,
Peter J.
Roth‡
ab,
Craig E.
Buckley
c and
Andrew B.
Lowe
*ab
aNanochemistry Research Institute (NRI), Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia. E-mail: andrew.b.lowe@curtin.edu.au
bDepartment of Chemistry, Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia
cDepartment of Physics and Astronomy, Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia
dCurtin Water Quality Research Centre, Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia
eFacultad del Mar y Medio Ambiente, Universidad del Pacifico, Guayaquil, Ecuador
fCentre for Microscopy, Characterisation and Analysis (CMCA), University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia
First published on 8th March 2016
Abstract
The synthesis and characterisation of soft matter nanoparticles based on AB diblock copolymers of oligo(ethylene glycol)methyl ether methacrylate (OEGMA) with 3-phenylpropyl methacrylate (PPMA) is described. Reversible addition–fragmentation chain transfer dispersion polymerization formulations that result in polymerization-induced self-assembly (RAFTDP-PISA) in methanol were utilized to access a range of poly(OEGMA-b-PPMA) (p(OEGMA-b-PPMA)) nanoparticles with the sphere-to-worm-to-vesicle order–order transitions being readily observed with increasing average degree of polymerization (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n) of the pPPMA block for a fixed n of 28 for the pOEGMA block. Similarly the effect of total copolymer concentration on the resulting nanoparticle morphology is also demonstrated whereby we highlight how tuning of worm micelle diameters can be accomplished simply by varying the concentration of a formulation. The block copolymer nanoparticles were characterized by size exclusion chromatography (SEC), 1H NMR spectroscopy, transmission electron microscopy (TEM) and small angle X-ray scattering (SAXS). Additionally, we report the first examples utilizing 3D electron tomography and in situ atomic force microscopy (AFM) in methanol as convenient and powerful complementary techniques for the characterization of the resulting soft matter nano-objects with an emphasis on the direct visualization of worm nanoparticles.
n) of the pPPMA block for a fixed n of 28 for the pOEGMA block. Similarly the effect of total copolymer concentration on the resulting nanoparticle morphology is also demonstrated whereby we highlight how tuning of worm micelle diameters can be accomplished simply by varying the concentration of a formulation. The block copolymer nanoparticles were characterized by size exclusion chromatography (SEC), 1H NMR spectroscopy, transmission electron microscopy (TEM) and small angle X-ray scattering (SAXS). Additionally, we report the first examples utilizing 3D electron tomography and in situ atomic force microscopy (AFM) in methanol as convenient and powerful complementary techniques for the characterization of the resulting soft matter nano-objects with an emphasis on the direct visualization of worm nanoparticles.
Introduction
The ability of an AB diblock copolymer, when placed in a selective solvent, to undergo self-directed assembly yielding nano-objects of various morphologies including, but not limited to, spheres,1 worms,2 disks,3 vesicles4 and more complex species is well-documented.5–9 Historically, such assemblies have been obtained by first preparing well-defined parent copolymers under homogeneous conditions followed by a processing step inducing self-assembly. Examples of such post-polymerization processing approaches include step wise dialysis or simple dilution with a selective solvent. While perfectly valid, these approaches can be time consuming, at least in the case of dialysis, and typically result in the generation of copolymer nano-object solutions of low concentration (≤1.0 wt% is common).Recently there has been significant academic interest in polymerization-induced self-assembly (PISA)10 – a process by which block copolymer formation and self-assembly occur in situ, thus negating the need for a post-polymerization processing step. Of particular relevance here is the recent development of reversible addition–fragmentation chain transfer dispersion polymerization (RAFTDP) formulations that result in PISA (RAFTDP-PISA).11–14 RAFTDP-PISA offers several significant advantages over the more traditional route for accessing polymeric nanoparticles. Firstly, and arguably most importantly, the process facilitates access to nano-objects with a range of morphologies in both mixed and pure phases including the often difficult to obtain worm, or cylindrical, micellar species; secondly, such nanoparticles can be prepared at concentrations significantly higher than those by the more traditional post-polymerization routes with formulations conducted at 40–50 wt% solids being readily achieved; thirdly, significant increases in the rate of polymerization, and hence nanoparticle formation, can be observed compared to homogeneous systems and this is especially relevant in aqueous-based formulations, although has also been observed in alcoholic-based formulations under microwave assisted conditions,15 and finally, from a practical viewpoint, it is no more difficult to execute than a conventional RAFT radical polymerization and is thus readily adopted and implemented.
Indeed, there now exists an impressive body of work highlighting various versatile formulations yielding nanoparticles possessing a variety of functionality and properties, with much of the work coming from the laboratories of Armes,16–21 Pan13,22–27 and Zhang.28–34 Zheng and Pan35 reported the first example of a RAFTDP-PISA system based on a polystyrene macro-CTA that was utilized in the subsequent block copolymerization of 4-vinylpyridine (4VP) in cyclohexane (a non-solvent for poly4VP) to give spherical micelles. Following this seminal contribution, a number of groups have examined the general RAFTDP-PISA approach for the preparation of soft matter nanoparticles of variable morphology in a range of media including polar (alcohols and water most commonly)11,18,20,23,24,36–57 and non-polar (typically alkanes)16,58–65 solvents as well as more exotic media such as super critical CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 66–70 and ionic liquids.71 Summaries of the current state-of-the-art can be found in several recent mini-review papers.10,11,14,37,72,73
66–70 and ionic liquids.71 Summaries of the current state-of-the-art can be found in several recent mini-review papers.10,11,14,37,72,73
We also have a fundamental interest in RAFTDP-PISA formulations and have developed a particular interest in methacrylic aromatic comonomers such as 2-phenylethyl methacrylate50 and 3-phenylpropyl methacrylate49 (PPMA) as the core forming species. The latter species, PPMA, is particularly attractive given its sub-ambient glass transition temperature (Tg).49 Typically we have employed macro-CTAs based on poly[2-(dimethylamino)ethyl methacrylate] in, predominantly, alcoholic49,50,56,57 solvents but have also utilized poly(stearyl methacrylate) macro-CTAs in non-polar media and specifically in n-tetradecane64 and n-octane.65 Extending our work on alcoholic PPMA-based formulations herein we describe the synthesis and characterisation of soft matter nanoparticles based on poly[oligo(ethylene glycol)methyl ether methacrylate] (pOEGMA) with PPMA as the comonomer. Basic features of the p(OEGMA-PPMA) block copolymer system are detailed and characterisation is accomplished utilizing size exclusion chromatography (SEC), NMR spectroscopy, transmission electron microscopy (TEM) and small angle X-ray scattering (SAXS). Additionally, and emphasizing the characterization of worm nanoparticles, we describe the first examples of tomographic imaging via 3D-TEM as well as in situ atomic force microscopy (AFM) in MeOH. We highlight how these latter two techniques are both powerful and yield complementary morphological information.
Experimental
All reagents were purchased from the Aldrich Chemical Company at the highest available purity and used as received unless noted otherwise. 3-Phenylpropyl methacrylate (PPMA) was purchased from Monomer-Polymer and Dajac Labs. 2,2′-Azobis(isobutyronitrile) (AIBN) was purified by recrystallization (twice) from methanol and then stored in a freezer until needed. Oligo(ethylene glycol)methyl ether methacrylate (OEGMA) (average![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 300) and PPMA were purified by passage through a basic Al2O3 column to remove the inhibitor and acidic impurities prior to use. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was prepared according to a procedure described elsewhere.74
n = 300) and PPMA were purified by passage through a basic Al2O3 column to remove the inhibitor and acidic impurities prior to use. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was prepared according to a procedure described elsewhere.74
Synthesis of poly[oligo(ethylene glycol)methyl ether methacrylate] (pOEGMA) via RAFT homopolymerization
Below is a general procedure for the RAFT homopolymerization of OEGMA mediated by CPADB.A solution containing OEGMA (10.0 g, 3.33 × 10−2 mol), CPADB (1.86 × 10−1 g, 6.67 × 10−4 mol), AIBN (2.18 × 10−2 g, 1.33 × 10−4 mol), and acetonitrile (30 mL) was added to a reaction vial equipped with a magnetic stir bar. The reaction vessel was sealed and the reaction mixture purged with argon for 30 min prior to being placed in a preheated oil bath at 70 °C. Polymerization was allowed to proceed for 4 h after which it was halted by exposure to air while cooling in an ice/water bath. The pOEGMA homopolymer(s) were isolated by precipitation into a mixture of diethyl ether and petroleum spirit (1:1, v/v) followed by filtration and dried overnight prior to NMR spectroscopic and SEC analysis.
RAFT dispersion copolymerization of PPMA with pOEGMA in alcoholic media
Below is a general procedure for the RAFTDP of PPMA with a polyOEGMA28 (pOEGMA28) macro-CTA in methanol. All RAFTDPs were performed in a similar fashion.A solution containing PPMA (1.65 × 10−1 g, 8.07 × 10−4 mol), AIBN (3.78 × 10−4 g, 2.30 × 10−6 mol), pOEGMA28 macro-CTA (1.0 × 10−1 g, 1.15 × 10−5 mol) and anhydrous methanol (1.34 mL) was added to a vial equipped with a magnetic stir bar. The reaction vessel was sealed and the solution purged with argon for 15 min prior to being placed in a preheated oil bath at 70 °C. Polymerization was allowed to proceed for 24 h after which it was halted by exposure to air while cooling in an ice/water bath. Block copolymer was isolated by precipitation into a large excess of diethyl ether and petroleum spirit mixture (1:1, v/v) followed by filtration and dried overnight prior to NMR spectroscopic and SEC analyses.
Size exclusion chromatography (SEC)
SEC analysis was performed on a Shimadzu modular system consisting of a 4.0 mm × 3.0 mm Phenomenex Security Guard™ Cartridge guard column and two linear phenogel columns (103 and 104 Å pore size) in tetrahydrofuran (THF) operating at a flow rate of 1.0 mL min−1 and 40 °C using an RID-20A refractive index detector, an SPD-M20A prominence diode array detector and a miniDAWN TREOS multi-angle static light scattering (MALLS) detector. The system was calibrated with a series of narrow molecular weight distribution poly(methyl methacrylate) standards with molecular weights ranging from 2.8 to 220 kg mol−1. Chromatograms were analysed using LabSolutions SEC software.Nuclear magnetic resonance (NMR) spectroscopy
NMR spectra were recorded on a Bruker 400 MHz spectrometer. 1H NMR spectra were recorded in deuterated chloroform (CDCl3). Residual CHCl3 (δ = 7.26 ppm) was utilized as the internal reference signal.Dynamic light scattering (DLS)
DLS measurements were performed on a Malvern Zetasizer Nano Series instrument (laser power = 4 mW, wavelength = 633 nm, detection angle = 173°) at 25 °C. For sample preparation, 50 μL of the parent RAFTDP solution was diluted with 1.45 mL of methanol or ethanol and the solution stirred for 10 min prior to filtration through 0.45 μm PTFE filters. For temperature-dependent DLS analysis, samples were heated and allowed to equilibrate at each temperature for at least 10 min prior to measurements.Transmission electron microscopy (TEM)
2-Dimensional (2D) TEM analyses were conducted on a JEOL 2100 transmission electron microscope operated at 120 kV. Samples for analysis were prepared by the slow addition of 50 μL of the parent RAFTDP solution to 1.45 mL of methanol under stirring. The copolymer solution (0.7 wt%) was dropped onto the top of a copper grid (ProSciTech) or a carbon-coated copper grid (SPI Suppliers) and allowed to contact for 1 min. Excess solution was wicked off using a filter paper. To stain the nano-objects, uranyl acetate (0.2 wt%) solution was soaked on the sample-loaded grid for 30 s and then carefully blotted to remove excess solution. Prior to TEM measurements, samples p(OEGMA28-b-PPMA66), p(OEGMA28-b-PPMA73) and p(OEGMA28-b-PPMA83) were dialyzed against methanol for several days to remove residual monomer. Average nanoparticle diameter and associated uncertainty were calculated for each polymer sample by averaging ∼30 particles from the TEM images using ImageJ software.For tomography measurements, polymer samples were prepared and stained as described above. Tomographic analyses were performed on a Titan G2 80-200 TEM/STEM instrument operated at 200 kV. The images were collected at sample-tilting angles ranging from −70° to 70° in 2° intervals. The series of images were reconstructed into a 3D image based on the filtered back projection method.
Small angle X-ray scattering (SAXS)
SAXS experiments were performed at the Australian Synchrotron on the SAXS/WAXS beamline. Measurements were made with a monochromatic X-ray beam (λ = 0.11271 nm) illuminating samples positioned 7.2 m from a Pilatus 1M detector. The SAXS patterns covered a scattering vector range of 0.01 nm−1 < q < 1.17 nm−1 where q = (4πsin(θ)/λ) defines the magnitude of the scattering vector and θ is half the scattering angle. Wet samples were mounted between Kapton tape on a frame with approximately 2 mm thickness. The 2-D scattering patterns were background subtracted and reduced on site using ScatterBrain and the data put onto an absolute intensity using a glassy carbon target as a secondary standard. Three samples were selected for measurement: p(OEGMA28-b-PPMA36), p(OEGMA28-b-PPMA69), and p(OEGMA28-b-PPMA71) at 10 wt%. To analyse the data the SASfit software package was used to model different form factors for the different nanoparticle phases.75
Atomic force microscopy (AFM)
AFM characterisation of worm nanoparticles was performed on a Bruker Dimension Icon system. Specifically, in situ AFM imaging was conducted in tapping mode in methanol. The images were acquired with SNL probes (silicon nitride cantilever, spring constant: 0.35 N m−1, uncoated silicon tip, maximum tip radius: ∼12 nm, Bruker, USA) at a scan rate of 0.5 Hz at 512 × 512 data points per image, and at room temperature. The frequency of the cantilever in methanol was approximately 83 kHz. The images were processed by Bruker Nanoscope Analysis 1.50 software. Sample preparation was conducted as follows. Worm nanoparticles were adsorbed to mica surface following the layer-by-layer protocol using iron oxide as an intermediate layer.76,77 Briefly, iron oxide precipitation was performed by increasing the pH of a 10 mM FeCl3 solution to 7 by small additions of 0.1 M NaOH solution. Then, 100 μL of iron oxide solution was pipetted on a 1 cm2 freshly-cleaved muscovite mica surface (V-1 quality, Electron Microscope Sciences, USA) and allowed to coat for 10 min. After careful rinsing with ultrapure doubled-deionized water (DDI, 18 MΩ cm resistivity, Millipore, USA), 20 μL of the worm nanoparticle solution was pipetted on the iron oxide-coated mica surface. Subsequently, 200 μL of methanol was pipetted on the substrate to prevent sample desiccation. The worm nanoparticles sample was immediately imaged after preparation. Due to high evaporation at room temperature, additional methanol solution was constantly injected to the sample stage using a 1000 μL pipette.Results and discussion
The general synthetic approach to the poly[oligo(ethylene glycol)methyl ether methacrylate-b-3-phenylpropyl methacrylate] (p(OEGMA-b-PPMA)) copolymers and their corresponding nanoparticles is given in Scheme 1. | ||
| Scheme 1 General approach for the preparation of the poly[oligo(ethylene glycol)methyl ether methacrylate-block-3-phenylpropyl methacrylate]-based nanoparticles by methanolic RAFT dispersion polymerization with polymerization-induced self-assembly. | ||
Initially, OEGMA (average n = 300) was homopolymerized under typical homogeneous RAFT conditions in MeCN at 70 °C with 4-cyanopentanoic acid dithiobenzoate (CPADB) as the RAFT chain transfer agent (CTA) and AIBN as the source of primary radicals for a target average degree of polymerization (n) at quantitative conversion of 50. Halting the homopolymerization at intermediate conversion yielded a well-defined polyOEGMA (pOEGMA) macro-CTA with an SEC-measured n of 5900 and corresponding dispersity (ĐM = w/n) of 1.06, and an NMR-determined n of 28 (pOEGMA28). This translates to an absolute molecular weight of ca. 8700 and OEGMA monomer conversion of 56%.
With a pOEGMA28 homopolymer (macro-CTA) available we next evaluated the RAFT dispersion polymerization (RAFTDP) of 3-phenylpropyl methacrylate (PPMA) in MeOH. Based on our previous work with PPMA in alcoholic RAFTDP formulations we anticipated that this would result in the in situ formation of p(OEGMA-b-PPMA) nanoparticles, of tuneable morphology, as a result of polymerization-induced self-assembly (PISA). In the first series of polymerizations we focused on the preparation of AB diblock copolymers with a fixed n of the pOEGMA block (pOEGMA28) while systematically varying the n of the pPPMA block for a fixed copolymer concentration of 20 wt%.
In general, all block copolymerizations proceeded smoothly with PPMA conversions generally ≥90% yielding AB diblock copolymers with compositions spanning the range p(OEGMA28-b-PPMA36)–p(OEGMA28-b-PPMA71). In contrast, copolymers with targeted compositions (at quantitative conversion) of pOEGMA:pPPMA of 28:125 and 28:133 both reached only 62% conversion during the 24 h polymerization period although still yielded highly asymmetric block copolymers with final pOEGMA:pPPMA compositions of 28:73 and 28:83 respectively. As expected, with increasing the n of the pPPMA block for a fixed n of the pOEGMA block both the NMR and SEC-measured molecular weights increased systematically while all measured block copolymer ĐM's were relatively low at ≤1.26, Table 1. Fig. 1 shows representative normalized SEC traces (refractive index signal) for the pOEGMA28 macro-CTA along with examples of the p(OEGMA28-b-PPMAx) copolymers.
n) as measured by NMR spectroscopy and SEC, their dispersities (ĐM), average nanoparticle hydrodynamic diameters as determined by DLS, the DLS polydispersity (μ2/Γ2) and the nanoparticle morphology as observed by TEM
| p(OEGMA-b-PPMA) compositiona | [PPMA]/[macro-CTA] | PPMA% conv.a |
n,NMR (n,SEC) |
Đ M b | Hydrodynamic diameterc (nm) | μ 2/Γ2c | TEM morphologyd |
|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy. b As determined by size exclusion chromatography in THF at a flow rate of 1.0 mL min−1. c As measured by dynamic light scattering. d S = spheres, W = worms, V = vesicles. | |||||||
| p(OEGMA28-b-PPMA36) | 40 | 95 | 16000 (12800) |
1.22 | 10.1 | 0.08 | S |
| p(OEGMA28-b-PPMA47) | 50 | 91 | 18300 (14100) |
1.22 | 11.6 | 0.07 | S |
| p(OEGMA28-b-PPMA57) | 58 | 90 | 20300 (15700) |
1.24 | 13.1 | 0.07 | S |
| p(OEGMA28-b-PPMA58) | 60 | 90 | 20500 (15900) |
1.23 | 15.8 | 0.12 | S + W |
| p(OEGMA28-b-PPMA69) | 70 | 96 | 22800 (18100) |
1.22 | 105.5 | 0.24 | W |
| p(OEGMA28-b-PPMA71) | 75 | 95 | 23200 (18400) |
1.26 | 363.3 | 0.51 | W |
| p(OEGMA28-b-PPMA73) | 125 | 62 | 23600 (19000) |
1.22 | 161.1 | 0.22 | W + V |
| p(OEGMA28-b-PPMA83) | 133 | 62 | 25200 (23400) |
1.18 | 79.8 | 0.64 | W + V |
 | ||
| Fig. 1 Representative SEC traces of the pOEGMA28 macro-CTA and the resulting p(OEGMA28-b-PPMAx) copolymers. | ||
While the measured ĐM's of the macro-CTA and resulting AB diblock copolymers were generally acceptable we did, in all cases, observe shoulders in the SEC traces at lower retention times for these materials. This is indicative of the presence of high molecular weight impurities resulting from polymeric radical–radical coupling reactions and is not uncommon in such RAFT (co)polymerizations taken to near-quantitative conversions. While the presence of such species is detrimental if the primary research goal is to prepare well-defined p(OEGMA-PPMA) block copolymers it is not necessarily problematic in PISA formulations where the principle aim is the preparation of nanoparticles with tuneable morphology.
Following NMR and SEC analysis all block copolymers were characterised using a combination of dynamic light scattering (DLS) and transmission electron microscopy (TEM). Fig. 2 shows representative high resolution TEM images of the p(OEGMA28-b-PPMAx) nano-objects formed in the above series of experiments. In the case of the near symmetric species, p(OEGMA28-b-PPMA36) (Fig. 2A), a pure spherical phase was observed that exhibited a narrow size distribution with the nanoparticles having an average TEM diameter of 17.3 ± 3.4 nm. This is in reasonable agreement with the DLS measured hydrodynamic diameter (Dh) of 10.1 nm (corresponding DLS polydispersity, μ2/Γ2, of 0.08). Increasing the n of the pPPMA block to 50 and 57 also resulted in the formation of pure spherical phases whose DLS measured sizes increased slightly to 11.6 (μ2/Γ2 = 0.07) and 13.1 nm (μ2/Γ2 = 0.07) respectively, see Table 1. Interestingly, an increase in the calculated n of the pPPMA block by 1 to 58 resulted in the formation of a mixed phase, Fig. 2B, consisting predominantly of spheres (average TEM diameter of ca. 18 nm) along with oligomeric worms formed from the 1D coalescence of approximately 3–4 spherical particles, vide infra. This average block copolymer composition (p(OEGMA28-b-PPMA58)) is clearly situated at the sphere-worm order–order transitional boundary. A further increase in the n of the pPPMA block to 69 resulted in a pure phase consisting of worm nano-objects, Fig. 2C. These worm nanoparticles are much better developed than those in Fig. 2B being significantly longer (on the order of several hundreds of nanometres) and with uniform widths of 17.2 ± 3.2 nm that agrees extremely well with the value as determined by small angle X-ray scattering (SAXS) experiments, vide infra. In the case of p(OEGMA28-b-PPMA71) a predominantly worm phase is observed, Fig. 2D, although the wall-to-wall distance is significantly greater (28.8 ± 3.6 nm) than that observed for the worm nanoparticles in 2C, but also agrees very well with the corresponding SAXS measurements for this sample. Fig. 2E shows essentially identical characteristics to that observed for Fig. 2D which is not surprising given the similarity of the block copolymer compositions. Finally, in Fig. 2F we observed the presence of a mixed phase consisting of worms and polymersomes with the latter being of a rather broad size distribution. Such sequential order–order transitions are consistent with previous reports from our group and the generally accepted effect of systematically increasing the n of the solvophobic block for a fixed solvophilic block, of appropriate length, at a fixed concentration, i.e. increasing the relative volume fraction of the solvophobic block for an essentially fixed volume fraction of the solvophilic block.
 | ||
| Fig. 2 Representative high resolution TEM images of the nanoparticles formed in situ in the methanolic RAFT dispersion polymerization synthesis of p(OEGMA28-b-PPMAx) copolymers at 20 wt% for a fixed pOEGMA n of 28 and variable pPPMA n. (A) p(OEGMA28-b-PPMA36); (B) p(OEGMA28-b-PPMA58); (C) p(OEGMA28-b-PPMA69); (D) p(OEGMA28-b-PPMA71); (E) p(OEGMA28-b-PPMA73), and (F) p(OEGMA28-b-PPMA83). | ||
SAXS is a powerful characterisation technique utilized here to obtain complementary information regarding the nanoscale morphology of selected examples of the p(OEGMA-PPMA) block copolymer nanoparticles. One key feature of SAXS experiments that makes it particularly useful for characterising nanoscale objects is that the obtained scattering data is averaged over millions of particles illuminated by the X-ray beam (vs. several hundred, or perhaps thousands, in the case of direct imaging such as in the TEM data presented in Fig. 2) and the results are, arguably, a more accurate reflection of the bulk sample. However, the representative TEM images do give a good indication of the expected morphology and is thus useful, as a starting point, for modelling of SAXS scattering data. The intensity measured in a SAXS experiment is the result of a number of different processes and interactions of the incident X-ray beam and the sample. In general, the intensity is considered a combination of three contributing aspects of the sample; the form factor (shape of the particles), the structure factor (the interaction between particles), and the size distribution (size of the particles). Ideal SAXS experiments minimize contributions from the structure factor by employing dilute samples, and size distribution contributions (monodisperse samples are preferred) so that data analysis can focus on the form factor giving accurate morphological information.78 Indeed, when measurements are made on a dilute solution of scattering particles the structure factor can be neglected.
Since we chose to evaluate 10 wt% samples, i.e. concentrated nanoparticle solutions the inter-particle interactions are significant and as such cannot be neglected in the modelling process and so a structure factor has been included. Also we note, that high concentrations, and associated inter-particle interactions, suppress scattering at low q. As such extracting relevant information at low q (the Guinier region) regarding morphology is extremely difficult while concentration tends to have little effect on higher q values.79 For concentrated polymer solutions it has been proposed that the structure factor can be calculated from the Random Phase Approximation (RPA).80 To take into account the size distribution of the particles a log-normal functional was included to describe micelle dimensions for all models.
Initially, the p(OEGMA28-b-PPMA36) sample was analysed, Fig. 3. In this model the form factor for a sphere was used with the RPA structure factor as well as a term for an exponential mass fractal (see ESI†). This term for the mass fractal was included to account for the self-similar structure of the polymer at larger sizes (shown by the sharp gradient at low q).
 | ||
| Fig. 3 Raw and fitted data for the SAXS analysis of the p(OEGMA28-b-PPMA36) nanoparticles at 10 wt% in methanol. The raw data was modelled with SASfit. | ||
We observed an excellent correlation of the raw data and the modelled taking into account the high concentration and need to include an RPA structure factor in the analysis. Utilizing the SASfit least squares fitting routines, the model was refined to physical values associated with the spherical micelles. The model indicates that the average sphere radius is 9.5 ± 0.1 nm giving an average nanoparticle SAXS diameter of 19.0 ± 0.2 nm. This value is in excellent agreement with the average size determined by TEM of 17.3 ± 3.4 nm. We also observe a correlation peak in the data located at q = 0.283 ± 0.005 nm−1. This indicates that there is a local ordering of the spherical nanoparticles in solution with the average distance between the centre of one sphere to another being 22.2 ± 0.5 nm.
We next examined the p(OEGMA28-b-PPMA69) sample which TEM indicates, Fig. 2C, has a pure cylindrical, or worm-like, morphology. Fig. 4 shows the experimentally obtained and modelled data for this sample analysed at 10 wt% in methanol.
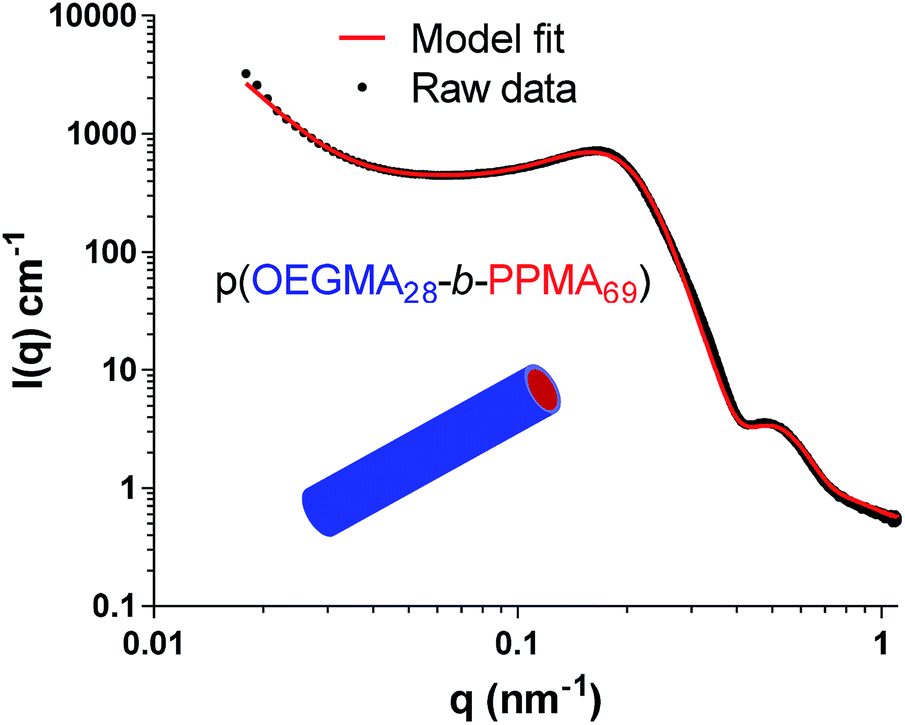 | ||
| Fig. 4 Raw and fitted data for the SAXS analysis of the p(OEGMA28-b-PPMA69) nanoparticles at 10 wt% in methanol. The raw data was modelled with SASfit. | ||
The raw data for these worm like micelles was modelled using a form factor developed specifically for worm-like aggregates and implemented into SASfit (see ESI† for further details)81 and the RPA structure factor and log-normal distribution implemented. Similar to the data for the p(OEGMA28-b-PPMA36) spherical species, a correlation peak is observed in the data and included in the model along with a mass fractal term.
The model gives good agreement around the local maxima at approximately q = 0.5 nm−1 – this feature in SAXS data is associated with a worm micelle cross-sectional size,63 and in this case the diameter as refined in the model for an average worm micelle width of 19.8 ± 0.4 nm. Again, this value agrees extremely well with the calculated average width of 17.2 ± 3.2 nm as determined by TEM, Fig. 2C. In this data, the correlation peak is located at q = 0.171 ± 0.005 nm−1, giving a distance of 36.7 ± 0.5 nm between the centres of interacting worm nanoparticles. Similar observations were made for the p(OEGMA28-b-PPMA71) worm nanoparticles and the data is presented in the ESI.†
Having successfully characterised the p(OEGMA-b-PPMA) nano-objects by TEM, selected samples by SAXS employing appropriate structure and form factors for spherical and worm micelles, the worm nanoparticles derived from p(OEGMA28-b-PPMA69), were further characterised by electron tomography (3D-TEM), a technique that has been demonstrated to be highly effective for the visualization of nano-sized objects, including soft matter species.82–84 This technique is likewise complementary to the traditional 2D TEM characterisation as well as the SAXS data. A series of 2D TEM images were obtained by tilting of the sample over an angular range of −70° to 70° in 2° increments about a single tilt axis. The acquired series of 2D images were then converted to the 3D image shown in Fig. 5. This is, we believe, the first example of a tomographic image of a pure polymeric-derived worm nanoparticle phase obtained via 3D-TEM.
 | ||
| Fig. 5 TEM-acquired tomographic image of the worm nanoparticles formed by the p(OEGMA28-b-PPMA69) copolymer highlighting the general volumetric uniformity of the worm nanoparticle species. | ||
While the uniformity, with respect to width, of the worm nanoparticles is clearly demonstrated by the 2D-TEM images, Fig. 2C, the tomographic image additionally confirms the general volumetric uniformity of the worm species. This is an important structural feature since, as we detail below, it is possible to prepare worm nanoparticles with differing sizes/widths simply by varying the synthesis conditions.
There are two additional features in Fig. 5 that are worth noting. Firstly, and highlighted in the yellow oval labelled 1, we can see that the worm nanoparticle is clearly composed of a series of aligned spherical species (although here the spherical cores are not fully coalesced) which confirms the general hypothesis that worm/cylindrical nano-objects are formed via the 1D fusion of spherical micelles, an order–order morphology transition mechanism that has been demonstrated recently with small molecule surfactants by Jensen and co-workers via SAXS.85 Secondly, worm nanoparticle overlap is more clearly observable. While we assume that the significantly lighter regions in Fig. 2C are due to nanoparticle overlap, as opposed to branching points, this is clearly evident in the tomographic image.
Atomic force microscopy (AFM) has evolved into a powerful technique for the characterisation of block copolymers in- and ex situ in either the unimeric or self-assembled states, although in situ measurements are still relatively uncommon.43,86–89 Exploiting this technique, and extending the range of suitable, complementary, characterisation techniques for such soft matter nano-objects, the worm nanoparticles formed by the p(OEGMA28-b-PPMA69) copolymer were characterised by in situ AFM in MeOH. This is the first reported example of in situ AFM characterisation of polymeric nanoparticles in MeOH and required the development of a new experimental approach for successful imaging (see Experimental section).
Height and phase images were collected simultaneously providing a correlation between topography and characteristics. Fig. 6A and B shows representative in situ images of the worm nanoparticles adsorbed on an iron oxide-coated mica surface with both the 3D (height) and 2D (phase) images shown. The height image shows the worm nanoparticles deposited on the substrate. The worm nanoparticles clearly have highly variable contour lengths (also apparent in the tomographic image) ranging from ca. 54 nm to >1 micron, with a mean contour length, as determined by probability density function analysis, Fig. 6E, of 251.8 nm. This is not uncommon and controlling the length distribution in such cylindrical micelles is extremely challenging. While the contour lengths are highly variable, both the diameter and heights are remarkably uniform, with statistical analysis giving an average diameter of 36.4 nm. This is somewhat larger than the calculated diameter from the TEM image in Fig. 2C of 17.2 ± 3.2 nm, as well as the modelled SAXS value, although we reiterate that analysis conditions via AFM are significantly different from those employed for both DLS and SAXS characterisation, with the sample set for the AFM analysis being much smaller than for TEM and SAXS. Additionally, our primary goal here is to report a new protocol for the effective in situ methanolic imaging of such worm nanoparticles via AFM as a new and powerful tool for characterising such species.
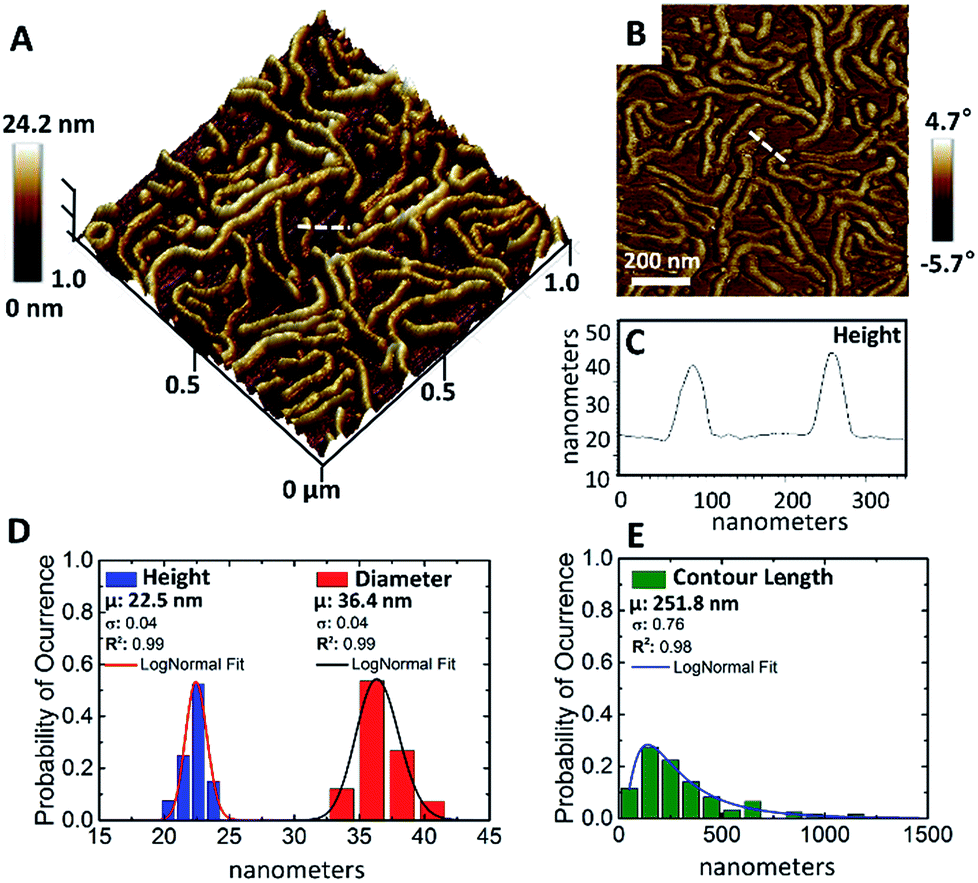 | ||
| Fig. 6 AFM height (A) and phase (B) micrographs (scan size = 1.0 μm × 1.0 μm) of p(OEGMA28-b-PPMA69) in methanol showing worm-like micelle assemblies (c = 7 × 10−3 wt%). The cross-sectional profile (C) was measured along the dashed line in image (A and B). Also shown is the statistical analysis to measure the height, cross-section diameter (D) and contour length (E) of worm-like nano-objects in methanol. | ||
Phase imaging records the phase shift signal, i.e. the delay in cantilever oscillation caused by energy dissipation involved in the contact between the tip and the worm nanoparticle sample, in tapping mode. The phase signal is particularly sensitive to variations in material properties, and phase contrast is one of the most commonly employed techniques for composition and mechanical characterisation of sample surfaces, composition, viscoelasticity, friction, topometric differences, elasticity, and other factors. However, understanding the contribution of each factor to the phase shift is not straightforward and cannot be easily separated from one another. The phase image, Fig. 6B, demonstrates how the cantilever oscillation was affected by interaction with the flat iron oxide-coated mica substrate and worm nanoparticles. Specifically, the phase image clearly shows regions with very different properties (i.e. clearly marked boundaries), with the worm nanoparticles being clearly visualized. This image is clear evidence of the difference in softness/hardness, as well as in chemical composition, between the soft matter worm nanoparticles and substrate. We are currently exploring this technique in more detail as a means of determining the nano-scale physical properties of such soft matter nano-objects.
Effect of block copolymer concentration for a fixed composition
One formulation variable that is known to have a direct effect on the resulting nanoparticle morphology is the total solids with, in general, increases in concentration resulting in more advanced or developed morphological states. Such behaviour is also common to RAFTDP-PISA systems and has been very clearly demonstrated in many of the phase profiles reported by Armes et al.18,49 as well as by others. To examine the effect of concentration we prepared three different p(OEGMA-PPMA) block copolymers with identical targeted pPPMAn's of 66 at three different concentrations, namely 10, 20 and 40 wt%. Based on the data presented above, a p(OEGMA28-b-PPMA66) block copolymer prepared at 20 wt% in MeOH would be expected to give nanoparticles with a pure worm morphology. Table 2 gives a summary of the key characteristics of the three block copolymers prepared.
:66 pOEGMA:pPPMA, the PPMA comonomer conversion, number average molecular weights (n) as measured by NMR spectroscopy and SEC, their dispersities (ĐM), average nanoparticle hydrodynamic diameters as determined by DLS, the DLS polydispersity (μ2/Γ2) and the nanoparticle morphology as observed by TEM
| p(OEGMA28-b-PPMAx) compositiona | Solids content (w/w) | PPMA% conv.a |
n,NMR (n,SEC) |
Đ M b | Hydrodynamic diameterc (nm) | μ 2/Γ2c | TEM morphology |
|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy. b As measured by size exclusion chromatography in THF at a flow rate of 1.0 mL min−1. c As determined by dynamic light scattering. | |||||||
| p(OEGMA28-b-PPMA66) | 10 | 57 | 22160 (16800) |
1.24 | 80.9 | 0.21 | Oligomeric worms |
| p(OEGMA28-b-PPMA66) | 20 | 88 | 22160 (17100) |
1.26 | 88.4 | 0.24 | Mostly worms |
| p(OEGMA28-b-PPMA67) | 40 | 93 | 22400 (17700) |
1.23 | 66.3 | 0.64 | ‘Fatter’ worms |
Targeting AB diblock copolymers with identical compositions via the RAFT radical polymerization process at such different concentrations can be problematic given the anticipated significant differences in polymerization kinetics. Gratifyingly, we were able to prepare three different block copolymers with essentially identical compositions as determined by 1H NMR spectroscopy, Table 2, with all resulting block copolymers also having similar dispersities. SEC traces for the three block copolymers are shown in Fig. 7A demonstrating the near-identical molecular weight distributions of the three samples. Interestingly, TEM analysis, Fig. 7B, demonstrated a clear effect of the synthesis conditions on the resulting nanoparticle morphology. In the 10 wt% formulation we observe a mixture of spheres and small ‘oligomeric’ worms clearly comprised of a small number of spherical species. In contrast, the 20 wt% formulation yielded a near-homogeneous worm phase in which the nanoparticles had end-to-end lengths of several hundred nanometers and wall-to-wall diameters of 17.2 ± 1.8 nm. Interestingly however, the worm nano-objects have a rather elongated appearance. This may be due to ionization of the carboxylic acid groups on the surface associated with the R group of the RAFT CTA, CPADB. Finally, in the case of the 40 wt% formulation, we observe another essentially pure phase but in this instance the average end-to-end distances appear more significant and the wall-to-wall diameters are clearly larger at 24.6 ± 1.6 nm. This highlights that in this p(OEGMA-PPMA) system accessing essentially pure worm phases with differing lengths and diameters is possible simply by controlling the concentration at which the particles are formed. Given that pure worm nano-object phases often only exist over a relatively narrow range of synthesis conditions this approach to accessing such species may offer some benefit versus simple tuning of block copolymer composition.
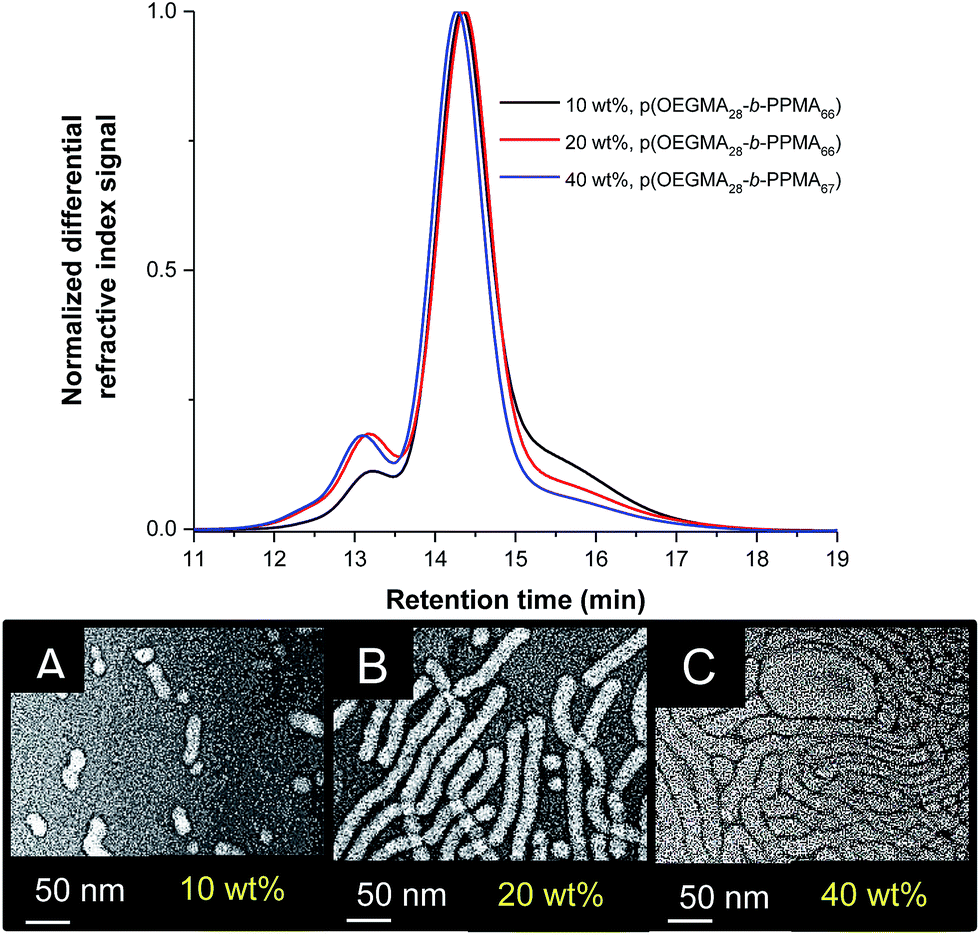 | ||
| Fig. 7 (A) Representative SEC traces (normalized refractive index signal) for the p(OEGMA-PPMA) block copolymers with targeted identical compositions prepared at different concentrations, and (B) representative TEM images of the resulting block copolymer nano-objects highlighting the effect of synthesis conditions on final nano-object morphology. (A) p(OEGMA28-b-PPMA66); (B) p(OEGMA28-b-PPMA66), and (C) p(OEGMA28-b-PPMA67). | ||
Conclusions
Herein we have described the synthesis and detailed characterisation of AB diblock copolymer nanoparticles based on poly[oligo(ethylene glycol)methyl ether methacrylate-b-3-phenylpropyl methacrylate] (p(OEGMA-b-PPMA)) formed in situ by the RAFTDP-PISA process in MeOH. By simply tuning final block copolymer composition and/or concentration it is possible to access the full range of common nanoparticle morphologies including spheres, worms and vesicles as determined by conventional 2D TEM experiments. The morphology of the spherical and worm nanoparticle species were also confirmed by advanced techniques including SAXS measurements performed at 10 wt%. Successful analysis required the incorporation of a structure factor into the data modelling based on the RDP model. Calculation of the cross-sectional diameter of the worm micelles yielded values entirely consistent with those obtained by TEM and DLS. We have also examined two, never before utilized, techniques for the further characterisation of the p(OEGMA-b-PPMA) worm nanoparticle species. Electron tomography (3D-TEM) was employed to generate a tomographic image of the worm species and highlighted the general volumetric uniformity of the worm nano-objects – this is an important feature given the ability to prepare worm objects with variable widths and heights simply by varying the concentration. Finally, we also developed a new method for the in situ visualization of the worm micelles by AFM. This allowed for the clear visualization of the nano-objects interacting with a mica surface, and likewise confirmed morphological features. Probability density function analysis allowed for a straightforward calculation of the average width, height and contour length of the micelles with the obtained data consistent with the complementary TEM and SAXS analysis. We are currently in the process of further examining the use of electron tomography and in situ alcoholic AFM for the direct visualization of soft matter nanoparticles.Acknowledgements
ABL thanks Curtin University for postdoctoral support for YP. ABL also thanks the ARC (FT110100046) for funding. The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. Part of this research was undertaken on the powder diffraction beamline at the Australian Synchrotron, Victoria, Australia.Notes and references
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- C. A. Dreiss, Soft Matter, 2007, 3, 956–970 RSC.
- L. Yin and M. A. Hillmyer, Macromolecules, 2011, 44, 3021–3028 CrossRef CAS.
- A. Eisenberg and D. E. Discher, Science, 2002, 297, 967–973 CrossRef PubMed.
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111–3118 RSC.
- J. Rodriguezhernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- E. T. Garrett, Y. Pei and A. B. Lowe, Polym. Chem., 2016, 7, 297–301 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Doudlas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS PubMed.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- C.-Q. Huang, Y. Wang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2011, 32, 1174–1179 CrossRef CAS PubMed.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- W.-M. Wan, X.-L. Sun and C.-Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed.
- X. Wang, J. Xu, Y. Zhang and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2452–2462 CrossRef CAS.
- P. Shi, C. Gao, X. He, P. Sun and W. Zhang, Macromolecules, 2015, 48, 1380–1389 CrossRef CAS.
- Y. Su, X. Xiao, S. Li, M. Dan, X. Wang and W. Zhang, Polym. Chem., 2014, 5, 578–587 RSC.
- Q. Li, C. Gao, S. Li, F. Huo and W. Zhang, Polym. Chem., 2014, 5, 2961–2972 RSC.
- Q. Li, F. Huo, Y. Cui, C. Gao, S. Li and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2266–2278 CrossRef CAS.
- S. Li, X. He, Q. Li, P. Shi and W. Zhang, ACS Macro Lett., 2014, 3, 916–921 CrossRef CAS.
- F. Huo, X. Wang, Y. Zhang, X. Zhang, J. Xu and W. Zhang, Macromol. Chem. Phys., 2013, 214, 902–911 CrossRef CAS.
- G. Zheng and C. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS.
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495–499 CrossRef CAS.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2014, 53, 1279–1283 CrossRef PubMed.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2013, 136, 1023–1033 CrossRef PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2010, 43, 2672–2675 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- C. Gao, S. Li, Q. Li, P. Shi, S. A. Shah and W. Zhang, Polym. Chem., 2014, 5, 6957–6966 RSC.
- Y. Pei, N. C. Dharsana, J. A. van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- M. Semsarilar, N. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2014, 6, 1751–1757 RSC.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2014, 47, 1664–1671 CrossRef CAS.
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- W. Zhou, Q. Qu, W. Yu and Z. An, ACS Macro Lett., 2014, 3, 1220–1224 CrossRef CAS.
- Y. Pei, N. C. Dharsana and A. B. Lowe, Aust. J. Chem., 2015, 68, 939–945 CrossRef CAS.
- Y. Pei, J.-M. Noy, P. J. Roth and A. B. Lowe, Polym. Chem., 2015, 6, 1928–1931 RSC.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J.-A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- L. Houillot, C. Bui, C. Farcet, C. Moire, J. A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CAS.
- J.-A. Raust, L. Houillot, M. Save, B. Charleux, C. Moire, C. l. Farcet and H. Pasch, Macromolecules, 2010, 43, 8755–8765 CrossRef CAS.
- M. Dan, F. Huo, X. Zhang, X. Wang and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1573–1584 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2014, 48, 236–244 CrossRef.
- Y. Pei, O. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC.
- A. M. Gregory, K. J. Thurecht and S. M. Howdle, Macromolecules, 2008, 41, 1215–1222 CrossRef CAS.
- H. Lee, E. Terry, M. Zong, N. Arrowsmith, S. Perrier, K. J. Thurecht and S. M. Howdle, J. Am. Chem. Soc., 2008, 130, 12242–12243 CrossRef CAS PubMed.
- M. Zong, K. J. Thurecht and S. M. Howdle, Chem. Commun., 2008, 5942–5944 RSC.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS PubMed.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- I. Bressler, J. Kohlbrecher and A. F. Thunemann, J. Appl. Crystallogr., 2015, 48, 1587 CAS.
- C. Aubry, L. Gutierrez and J. P. Croue, Water Res., 2013, 47, 3109–3119 CrossRef CAS PubMed.
- L. Gutierrez and T. H. Nguyen, Environ. Sci. Technol., 2012, 46, 8705–8713 CrossRef CAS PubMed.
- O. Glatter and O. Kratky, Small-angle X-ray Scattering, Academic Press, London, 1982 Search PubMed.
- V. M. Garamus, J. S. Pedersen, H. Kawasaki and H. Maeda, Langmuir, 2000, 16, 6431–6437 CrossRef CAS.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- A. L. Kholodenko, Macromolecules, 1993, 26, 4179–4183 CrossRef CAS.
- O. Ersen, C. Hirlimann, M. Drillom, J. Werckmann, F. Tihay, C. Pham-Huu, C. Crucifix and P. Schultz, Solid State Sci., 2007, 9, 1088–1098 CrossRef CAS.
- S. Kohjiya, A. Kato and Y. Ikeda, Prog. Polym. Sci., 2008, 33, 979–997 CrossRef CAS.
- S. Kohjiya, A. Katoh, J. Shimanuki, T. Hasegawa and Y. Ikeda, Polymer, 2005, 46, 4440–4446 CrossRef CAS.
- G. V. Jensen, R. Lund, J. Gummel, T. Narayanan and J. S. Pedersen, Angew. Chem., Int. Ed., 2014, 53, 11524–11528 CrossRef CAS PubMed.
- V. Percec, C.-H. Ahn, G. Ungar, D. J. P. Yeardley, M. Moller and S. S. Sheiko, Nature, 1998, 391, 161–164 CrossRef CAS.
- I. LaRue, M. Adam, M. Pitsikalis, N. Hadjichristdis, M. Rubinstein and S. S. Sheiko, Macromolecules, 2006, 39, 309–314 CrossRef CAS.
- K. Matyjaszewski, S. Qin, J. R. Boyce, D. Shirvanyants and S. S. Sheiko, Macromolecules, 2003, 36, 1843–1849 CrossRef CAS.
- G. Kreutzer, C. Ternat, T. Q. Nguyen, C. J. G. Plummer, J.-A. E. Manson, V. Castelletto, I. W. Hamley, F. Sun, S. S. Sheiko, A. Herrmann, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco and H.-A. Klok, Macromolecules, 2006, 39, 4507–4516 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04649e |
| ‡ Current address: Department of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, GU2 7XH, UK. |
| This journal is © The Royal Society of Chemistry 2016 |