
Polyoxometalate (POM) grafted grooved nanofibrous membranes for improved self-decontamination†
N. E. Allen*a,
S. K. Obendorfb and
J. Fanc
aCornell University, 37 Forest Home Dr, Ithaca, NY 14850, USA. E-mail: nea34@cornell.edu
bCornell University, 37 Forest Home Dr, Ithaca, NY 14850, USA. E-mail: sko3@cornell.edu
cCornell University, Forest Home Dr, Ithaca, NY 14850, USA. E-mail: jf456@cornell.edu
First published on 2nd September 2016
Abstract
Combining unique fiber morphologies with self-decontamination technologies can enhance degradation of toxic compounds on fibrous substrates. Co-continuous electrospinning was used to fabricate nanofibrous cellulose acetate membranes with unique, grooved fiber morphology due to the selective dissolution of polyethylene oxide. The grooved cellulose acetate membranes were deacetylated to form grooved cellulose membranes. A self-decontaminating polyoxometalate (POM)‡ (H5PV2Mo10O40)§ was synthesized and used to functionalize the grooved nanofibrous membranes as well as non-grooved nanofibrous membranes, pure cotton, 65/35 polyester/cotton fabrics and Tencel® peachskin fabric. The POM-treated substrates were exposed to methyl parathion, a simulant of the chemical warfare agent (CWA)¶ VX||, to evaluate the decontamination performance. It was found that the POM grafted, grooved nanofibrous cellulose membrane has significantly improved self-decontamination, owing to increased amount of POM grafted as a result of increased surface area. This work contributes to the use of novel fiber morphologies in breathable chemical protective apparel to achieve better CWA decontamination and comfort.
Introduction
Polyoxometalates (POMs) are heteropolyanions, which can be used to oxidize or hydrolyze chemical warfare agents. These reactions can degrade toxins into less harmful byproducts. When the oxidation reaction occurs, the POM is reduced. Oxygen or any oxidant in the system can then re-oxidize the POM to its original state; the self-decontaminating agent is regenerated to be used for multiple decontamination cycles.1In order to develop self-decontaminating chemical protective apparel, Hill's group2–5 and others6 have incorporated POMs into fibers and fabrics such as micro-/nanofibers, polyacrylic, nylon and cotton fabrics, and polyurethane sponges to create catalytic self-decontamination of volatile organics, air toxins, and chemical warfare agents.2,3,5,6 More recently, based on Hill's work, Lange and Obendorf showed that POMs can be incorporated into metal organic frameworks-another oxidization agent, at room temperature and grown on cotton to improve self-decontamination performance in protective clothing.7,8
Although POMs such as H5PV2Mo10O40 have been incorporated into cellulose substrates to degrade volatile organics and chemical warfare agents such as CEES,** it is still unclear how well they effectively degrade organophosphates.2,5,6 It is also desirable to increase the loads of self-decontaminating compounds in fibrous substrates to enhance the self-decontaminating performance without compromising the breathability and comfort of such materials.
It is therefore the objective of the present work to fabricate grooved nanofibrous membranes, functionalize them with polyoxometalates (POMs), and evaluate their self-decontamination performance in degrading toxic organophosphates. The increased surface area of grooved nanofibrous membranes in comparison with non-grooved nanofibrous membranes and conventional textile substrates is expected to increase the loads of POMs grafted on the fiber surfaces, resulting in enhanced self-decontamination performance.
Experimental
Materials
Cellulose acetate (Mn 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in powder form and poly(ethylene oxide) (Mv 200000) were obtained from Sigma-Aldrich, in addition to the solvents acetonitrile HPLC grade from EMD Chemicals, Inc and acetone from Mallinckrodt Chemicals, for electrospinning. BD Sterifill SCF 5 mL plastic syringes and a Harvard Apparatus PHD ULTRA electrospinning device in combination with a Gamma High Voltage Research voltage source were used to fabricate the fibrous membranes. 100% cotton plain-weave non-bleached swatches were obtained from Testfabrics, Inc. The Tencel® test fabric samples were obtained from Lenzing Corp. customer service. A standard solution of 0.1 N sodium hydroxide obtained from Sigma-Aldrich and ethanol obtained from Mallinckrodt Chemicals were used for the deacetylation process. H5PV2Mo10O40 was synthesized using sodium molybdate dihydrate from EMD Chemicals, Inc, sodium metavanadate from Sigma-Aldrich, sodium phosphate dibasic from Sigma-Aldrich, 0.5 N sulfuric acid from Sigma-Aldrich Fluka Analytical, and methanol HPLC grade from Sigma-Aldrich. Tin(II) 2-ethylhexanoate and hexamethylene diisocyanate (HMDI)†† were obtained from Sigma-Aldrich for membrane grafting. Toluene and hexane were obtained from Mallinckrodt Chemicals. The pesticide, methyl parathion, and its degradation product, methyl paraoxon, otherwise known as dimethyl-p-nitrophenylphosphate, were purchased from Chem Service Incorporated. p-Nitrophenol was purchased from Sigma-Aldrich Fluka Analytical.
000) in powder form and poly(ethylene oxide) (Mv 200000) were obtained from Sigma-Aldrich, in addition to the solvents acetonitrile HPLC grade from EMD Chemicals, Inc and acetone from Mallinckrodt Chemicals, for electrospinning. BD Sterifill SCF 5 mL plastic syringes and a Harvard Apparatus PHD ULTRA electrospinning device in combination with a Gamma High Voltage Research voltage source were used to fabricate the fibrous membranes. 100% cotton plain-weave non-bleached swatches were obtained from Testfabrics, Inc. The Tencel® test fabric samples were obtained from Lenzing Corp. customer service. A standard solution of 0.1 N sodium hydroxide obtained from Sigma-Aldrich and ethanol obtained from Mallinckrodt Chemicals were used for the deacetylation process. H5PV2Mo10O40 was synthesized using sodium molybdate dihydrate from EMD Chemicals, Inc, sodium metavanadate from Sigma-Aldrich, sodium phosphate dibasic from Sigma-Aldrich, 0.5 N sulfuric acid from Sigma-Aldrich Fluka Analytical, and methanol HPLC grade from Sigma-Aldrich. Tin(II) 2-ethylhexanoate and hexamethylene diisocyanate (HMDI)†† were obtained from Sigma-Aldrich for membrane grafting. Toluene and hexane were obtained from Mallinckrodt Chemicals. The pesticide, methyl parathion, and its degradation product, methyl paraoxon, otherwise known as dimethyl-p-nitrophenylphosphate, were purchased from Chem Service Incorporated. p-Nitrophenol was purchased from Sigma-Aldrich Fluka Analytical.
Methodology
:PEO mixture of solids was then added to a vial, and a 60/40 volume ratio of acetonitrile to acetone was added to make a 5 mL solution. The solution was sonicated for 30 min and then stirred at room temperature on a high speed for 1 h.10–13The solution was immediately loaded into a 5 mL plastic syringe for electrospinning. A syringe pump (PHD Ultra Pump; Harvard Apparatus) infused the polymer solution at a speed of 0.05 mL min−1 through a stainless steel needle (Hamilton) with a diameter of 0.016 in (0.41 mm) for 1 h. A 15 kV voltage was applied between the needle and an aluminum foil wrapped wire mesh (4 in2 surface) taped to a Teflon® collection plate that was 15 cm from the syringe needle tip. An electric field was generated by the voltage between the needle tip and the collection surface enabling electrospun fibers to be deposited on the plate forming a nonwoven, nanofibrous mat. Experiments were conducted at room temperature. The foil wrapped collection plate with the nanofibrous web composed of 60% cellulose acetate and 40% polyethylene oxide was then removed from the Teflon® plate, placed in the vacuum oven, and vacuum dried for a minimum of 8 h at room temperature.
The nanofibrous electrospun fiber mats were carefully peeled from the foil and stored under vacuum until future experiments, where they were referred to as non-grooved/non-extracted fibrous membranes. To fabricate the grooved/extracted fibrous membranes, the electrospinning process detailed previously was performed using a volume of 8.3 mL of the 60/40 CA:PEO polymer solution so that the grooved/extracted membranes would have almost the same weight per unit area as that of the non-grooved/non-extracted membranes after the removal of PEO. The resulting fiber mats were carefully peeled from the foil and stored under vacuum for 24 h before being rolled gently into a loose roll using tweezers and then placed in a thimble filter. The thimble filter was placed in the glassware depository to conduct a Soxhlet extraction at 80 °C using HPLC grade deionized water for 8 days to remove the PEO component from the 60/40 CA:PEO fiber mat. The extracted fibrous membranes were then removed from the thimble filter with tweezers and vacuumed dried at room temperature for 24 h.
A portion of the extracted fibrous cellulose acetate membranes was deacetylated with 0.05 N sodium hydroxide in ethanol for 2.5 h. The resulting membranes were vacuum dried for 24 h, and FTIR‡‡ analysis was used to confirm the complete conversion of cellulose acetate to cellulose.14,15 As shown in Table 1, all textile substrates used for testing were cut to a dimension of 3 cm × 3 cm; the weights were measured using an analytical balance, and the thicknesses were measured using digital calipers.
| Samples (3 cm × 3 cm) | n | Mean weight (g) | g m−2 | Mean thickness (mm) | ||
|---|---|---|---|---|---|---|
| Non-grooved/non-extracted 60/40 CA:PEO membranes |
5 | 0.055 | ±0.003 | 60.56 | 0.23 | ±0.020 |
| Grooved/extracted 60/40 CA:PEO membranes |
5 | 0.053 | ±0.004 | 58.67 | 0.23 | ±0.018 |
| Grooved/extracted & deacetylated cellulose membranes | 5 | 0.052 | ±0.002 | 57.33 | 0.22 | ±0.015 |
| 100% cotton Testfabrics plain weave | 5 | 0.116 | ±0.010 | 129.33 | 0.23 | ±0.005 |
| 100% Tencel® Testfabrics twill | 5 | 0.167 | ±0.004 | 185.56 | 0.27 | ±0.004 |
After characterization to confirm the HMDI surface functionalization, the specimen was placed in a 200 mL three neck flask that contained a 1% w/v grafting dispersion of H5PV2Mo10O40 in toluene under nitrogen purge and rapidly agitated at a temperature of 45 °C for 24 h. It is primarily the pendant oxygens on the molybdenum and vanadium geometries of the polyoxometalate that are thought to be responsible for the formation of physical linkages between the isocyanate and the POM.17,18 After 24 h, 100 mL of deionized water was added to the flask under high speed agitation using a stir bar for 1 h to prevent POM agglomeration on the surface of the fibrous substrates. The reaction continued to be held under nitrogen purge at a temperature of 45 °C. The specimen was removed and placed in a toluene bath for 3 h at RT and then three subsequent 24 h baths of deionized water. These baths were used to remove any unreacted POM from the grafted substrates. The specimen was then vacuum dried overnight at RT. This grafting procedure was performed on five of each of the textile substrates listed in Table 1, and these substrates were characterized using FTIR, SEM,|||| and TGA.
Membrane samples were characterized via FTIR in order to detect the loss of polyethylene oxide (post-Soxhlet extraction), completeness of the conversion of cellulose acetate to cellulose, and the presence of HMDI and/or polyoxometalates (post-functionalization). FTIR spectra for cellulose acetate and polyethylene oxide polymers were measured separately from each other in powder form and compared to the results found in the literature. FTIR spectra for HMDI (liquid form) and H5PV2Mo10O40 POM (powder form) were used as controls in order to interpret the FTIR peaks generated from the functionalized specimens. The membrane samples were sputter coated with palladium and gold in order to make the samples conductive and prepare them for SEM analysis on the Tescan Mira3 FESEM. The POM percent add-on was quantified by measuring the weight loss of each substrate before and after POM-treatment via TGA using a TA Instruments Q500 Thermogravimetric Analyzer. The surface areas of the membranes were determined using the Brunauer, Emmett, Teller method known as BET prior to the grafting process. This molecular gas adsorption technique employed multipoint krypton adsorption at −195 °C on a BET Sorptometer at Porous Materials, Inc in Ithaca, NY.
A calibration curve relating the concentrations of methyl parathion to the peak area given by the HPLC-DAD was replicated from the Lange and Obendorf7 procedure by making a 100 mg L−1 stock standard solution of methyl parathion in hexane. Methyl parathion was dissolved in hexane at the following concentrations: 0.25, 0.5, 5, 10, 25 mg L−1, and HPLC data from these specimens were used to determine a standard equation for the relationship between methyl parathion concentration and the HPLC-DAD peak area. The result of this equation was then multiplied by the known volume to obtain the mass of methyl parathion. Wavelength scans at 320 nm were used to determine the amount of methyl parathion for each specimen.
Standards for the expected degradation products of p-nitrophenol and methyl paraoxon (a.k.a. dimethyl-p-nitrophenyl phosphate) were analyzed using the HPLC-DAD to determine their retention times. A standard solution for p-nitrophenol was made at a concentration of 0.070 g L−1 in HPLC grade deionized water, and a standard solution for methyl paraoxon was made at a concentration of 0.4 g L−1 in hexane. These solutions were sonicated for 30 min prior to testing. After achieving complete dissolution, 1 mL of these solutions was taken with a syringe. A syringe filter (nylon Cameo II 25 mm) was attached, and the filtered solutions were transferred to the corresponding HPLC vial. These samples were analyzed on the HPLC-DAD using the same method as described above. Each time the degradation performance of the polyoxometalate grafted substrates was measured, these standard solutions of degradation products were made and analyzed on the HPLC-DAD so that degradation products resulting from the POM decontamination could be recognized by the retention times of the peaks.
A standard solution of 0.016 g L−1 methyl parathion in hexane was made and sonicated for 30 min. To determine the decontamination performance of the polyoxometalate functionalized substrates, each POM grafted specimen was vacuumed dried for 24 h at RT and then placed in individual sealed containers of 20 mL of the methyl parathion standard solution. 1 mL aliquots were withdrawn from each vial by a syringe, filtered (nylon Cameo II 25 mm HPLC syringe filter), and placed in 1.5 mL HPLC vials at varying time intervals between 1 h and 1 week (1 h, 2 h, 5 h, 10 h, 24 h, 48 h, 72 h, 168 h). These solutions as well as hourly aliquots of methyl parathion standard solution were then analyzed on the HPLC-DAD using the method detailed above. After one week (168 h), each specimen was then removed from the 20 mL vial and washed thoroughly in hexane, vacuum dried, and stored in a vacuum desiccator.
Results and discussion
Characterization of polyoxometalate H5PV2Mo10O40 synthesis
Fig. 1(a) shows the FTIR spectrum of the synthesized particles with absorption bands at 3462, 1650, 1160, 1060, 858, 775, and 600 cm−1. The strong, broad band at 1160 cm−1 correlates well with the central phosphate tetrahedral anion.28,31 The strong, broad band at 1060 cm−1 correlates closely with the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, P–O functional groups, and the stretching mode of VO, as well as peaks seen in the literature for PO43− and MoO3.18,22–27 A strong, broad peak at 858 cm−1 correlates closely with MoO3 compound spectra and also can be attributed to the M–O–M vibrations of a doubly bridging oxygen connecting two contiguous chains in the structure. This broad peak encompasses part of the region (722–840 cm−1) where these M–O–M vibrations occur when the bond angles of the complexes are greater than 140°.24,25,28,29
O, P–O functional groups, and the stretching mode of VO, as well as peaks seen in the literature for PO43− and MoO3.18,22–27 A strong, broad peak at 858 cm−1 correlates closely with MoO3 compound spectra and also can be attributed to the M–O–M vibrations of a doubly bridging oxygen connecting two contiguous chains in the structure. This broad peak encompasses part of the region (722–840 cm−1) where these M–O–M vibrations occur when the bond angles of the complexes are greater than 140°.24,25,28,29
 | ||
| Fig. 1 (a) FTIR% transmittance spectra of H5PV2Mo10O40 from experimental synthesis (b) SEM of H5PV2Mo10O40. | ||
Although there are similarities, the observed IR peaks of the experimentally synthesized POM have some differences from what has been reported in the literature particularly in terms of the peak intensity. Some of these differences may be due to residual lattice moisture while others indicate that the synthesized compound may be an isomer of PV2Mo10 or a lacunary form of PV2Mo9. These possibilities could result from pH fluctuation during any of the processes such as synthesis or substrate-functionalization. While pH was carefully monitored during these processes to achieve the desired products, it was also observed that these reactions, particularly POM synthesis, are very sensitive to minimal pH changes.18,22,23,26,28
SEM images and further ImageJ‡‡‡ analysis showed that POM particle size ranged from 0.5–5 μm.
Characterization of fibrous substrates
When the polymers cellulose acetate and polyethylene oxide were analyzed via FTIR spectroscopy in their solid phase, their absorption signature showed major peaks at 1740 and 1032 cm−1 for cellulose acetate and at 2879, 1090, and 841 cm−1 for the polyethylene oxide. The strong peak at 1740 cm−1 correlated with an aliphatic acetate ester functional group, while the strong, broad peak at 1032 cm−1 correlated with the secondary primary alcohol functional groups of cellulose.25,30Fig. 2 compares the FTIR spectra of both the (a) non-grooved/non-extracted and (b) grooved/extracted fibrous membranes confirming that the PEO was removed from the 60/40 CA:PEO membrane via the Soxhlet extraction using deionized water in the grooved fibers due to the significant decrease in the size of the peaks at 2879, 1090, and 843 cm−1, which are characteristic of PEO. The decrease in the size of the other peaks, specifically at 1740 and 1032 cm−1, represent the loss of some cellulose acetate during the extraction process.
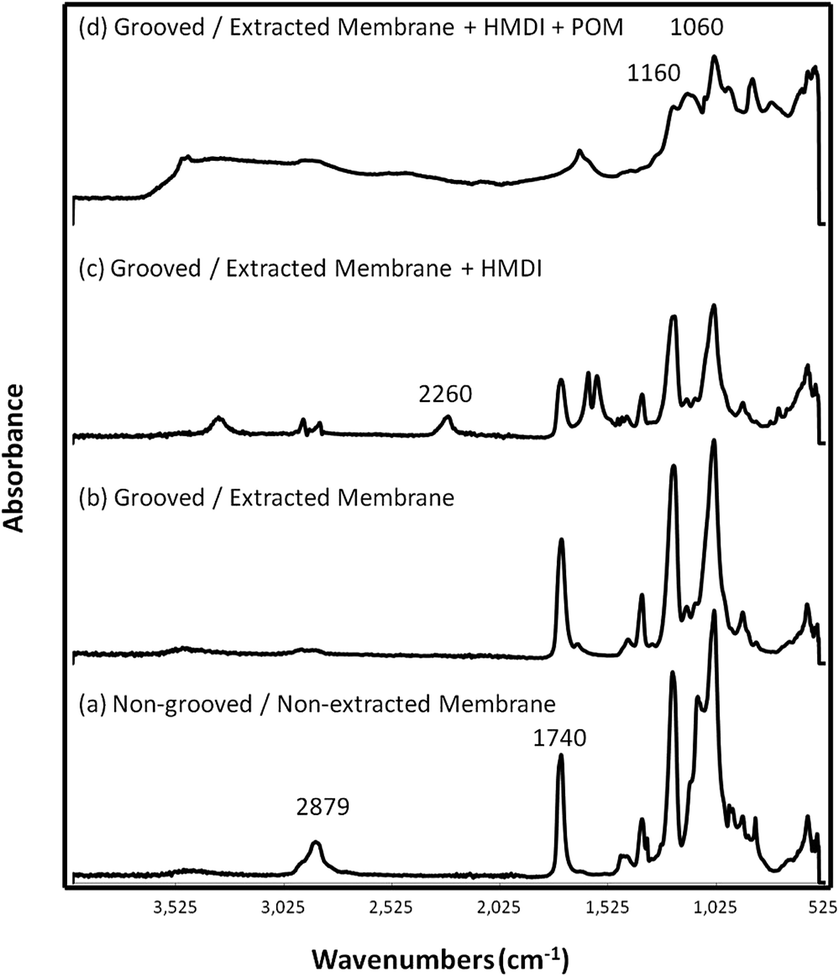 | ||
| Fig. 2 FTIR absorbance of the (a) non-grooved/non-extracted 60/40 CA:PEO fibrous membranes, (b) grooved/extracted cellulose acetate fibrous membranes, (c) grooved/extracted cellulose acetate fibrous membranes grafted with HMDI, and (d) grooved/extracted cellulose acetate fibrous membranes grafted with HMDI and the POM H5PV2Mo10O40. | ||
In comparing Fig. 3(a) of the non-grooved/non-extracted fibers to Fig. 3(b) of the grooved/extracted fibers, it is evident that the extraction of PEO led to the creation of grooved fiber morphology throughout the structure of the membrane. Fig. 3(a) shows that before extraction, the 60/40 CA:PEO fiber surfaces are smooth, while Fig. 3(b) shows more surface roughness. Achieving this grooved fiber morphology was critical to testing the hypotheses. ImageJ analysis of SEM Fig. 3(a) and (b) of the membranes showed that the fibers ranged from 1 to 2 μm in diameter both before and after extraction. Therefore, the grooved membranes had the same fiber diameter but greater surface roughness than the non-grooved fibers. Additional ImageJ analysis determined that the size of the grooves ranged from 0.01–0.8 μm.
 | ||
| Fig. 3 (a) Non-grooved/non-extracted 15% w/v 60/40 CA:PEO fibrous membranes (b) grooved/extracted cellulose acetate fibrous membranes after the Soxhlet extraction of polyethylene oxide. | ||
BET testing determined that the mean surface area of the non-grooved/non-extracted membranes was 0.03 m2 g−1 with a standard deviation of 0.01, while the mean BET surface area of the grooved/extracted membranes was 1.61 m2 g−1 with a standard deviation of 0.65 (Fig. 4). As expected, the surface area of the grooved membranes was higher than reported values for cellulose acetate fabrics (average nitrogen adsorption BET surface area of 0.38 m2 g−1) most likely due to the grooved fiber morphology created by the PEO extraction.31 The mean multipoint krypton adsorption BET surface area of the grooved and deacetylated cellulose membranes was 1.43 m2 g−1 with a standard deviation of 0.41 which is comparable to the surface area of the grooved cellulose acetate membranes indicating that the conversion of the cellulose acetate membranes to cellulose did not result in a significant difference in surface area. The surface area of the 100% cotton plain weave test fabric was 0.06 m2 g−1 with a standard deviation of 0.01, and the mean surface area of the Tencel® peachskin fabric was 0.17 m2 g−1 with a standard deviation of 0.02 (Fig. 4). One-way ANOVA statistical analysis of the BET surface area measurements via krypton gas adsorption of the non-grooved and grooved fibrous membranes showed that the difference in surface area between the grooved and non-grooved treatments was statistically significant with a p-value of 0.0001. A more conservative Wilcoxon/Kruskal–Wallis Test also showed that the increase in surface area for the grooved membranes was statistically significant with a p-value of 0.0197. This increase in surface area for the grooved fibers was critical to testing the hypotheses pertaining to an increased decontamination performance via increased fiber surface area for POM binding.
 | ||
| Fig. 4 Multipoint krypton adsorption BET surface area (m2 g−1) of the fibrous substrates. | ||
Characterization of H5PV2Mo10O40 treated fibrous substrates
When hexamethylene diisocyanate (HMDI) (liquid) was analyzed via FTIR, major peaks were observed at 2260, 1660, 1160, 1040, 860, and 573 cm−1. The spectrum of HMDI indicated several functional groups that are similar to that of the POM powder; however, it was apparent that a peak at 2260 cm−1 was unique only for HMDI as seen in Fig. 2(c). Examining the literature, it was found that nitrile groups can be identified from the FTIR spectrum in the triple bond region of 2300–2000 cm−1. For isocyanates, a type of alkyl nitrile functional group, FTIR absorption bands are typically seen in the 2275–2240 cm−1 region of the spectrum. A peak at 2260 cm−1 or 2240 cm−1 correlates to the presence of a saturated alkyl nitrile.25,30 This peak at 2260 cm−1 was used as the reference peak to confirm the presence of the HMDI, and the resulting spectrum was used as a standard of comparison when analyzing the POM grafted electrospun membranes.As discussed previously, 1160 and 1060 cm−1 were chosen as the defining peaks for the molybdovanadate POM since these peaks corresponded well to the P–O, Mo–O, and V–O functional groups as well as the M–O–M vibrations.18,22–27 Further evidence that the POM was present on the surface of the membranes included the presence of peaks and bands at 1650, 856, 768, and 600 cm−1.18,22–26,28,30,32
It was difficult to detect the full reaction of HMDI with POMs for the POM grafted membranes in Fig. 2(d) since the bands that resulted from the presence of the POM in the range of 2200–3600 cm−1 appeared to have coincided with the HMDI band at 2260 cm−1. The strong, broad bands seen at 1640, 2240, 3200, 3537, and 3590 cm−1 in the spectrum of the POM grafted HMDI functionalized membrane are likely due to residual lattice water commonly found in the structure of the POM.32 This observation can be correlated with the presence of the bands at 1640, 2240, 3200, 3537, and 3590 cm−1 in the spectrum of the POM powder control.
Fig. 5(a) shows images obtained via scanning electron microscopy of the grooved/extracted fibrous membranes prior to POM grafting, while Fig. 5(b) shows the grooved/extracted fibrous membranes after being grafted with the POMs with POM particles seen on the surface of the fibers where they formed a mono layer. No seeding was observed.
 | ||
| Fig. 5 (a) Grooved cellulose acetate fibrous membranes prior to POM grafting (b) grooved cellulose acetate fibrous membranes after POM grafting. | ||
Thermogravimetric weight loss analysis was used to measure the amounts of POM attached to the substrates (Fig. 6). The greatest POM percent add-on by weight of substrate occurred for the high surface area grooved electrospun cellulose acetate fibers (16.27% POM add-on) and grooved electrospun cellulose fibers (13.20% POM add-on).
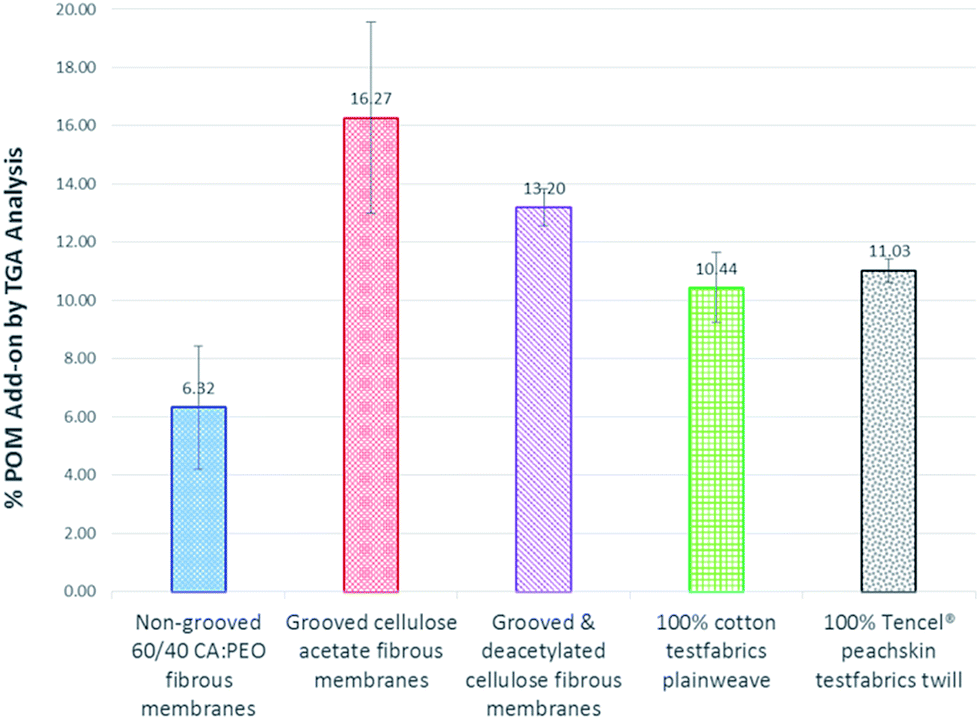 | ||
| Fig. 6 TGA weight loss analysis: amount of POM grafted to fibrous substrates. | ||
These amounts of POM percent add-on were significantly higher (p < 0.05) than those achieved for the lower surface area non-grooved electrospun cellulose acetate/polyethylene oxide fibers (6.32% POM add-on) and the 100% cotton fibers (10.44% POM add-on). These results correlate to the surface areas of the substrates indicating that increasing the amount of substrate surface area will increase the amount of achievable POM attachment up to a point. This relationship is illustrated in Fig. 7.
 | ||
| Fig. 7 Relationship between substrate surface area and percent POM loading. | ||
Decontamination of methyl parathion by POM grafted fibrous membranes/textile substrates
Degradation of methyl parathion was observed when POMs were grafted to fibrous substrates. No methyl parathion degradation was observed on non-POM grafted materials. Fig. 8 represents the decontamination performance of the POM grafted grooved cellulose acetate and non-grooved 60/40 CA:PEO membranes after 24 h. It shows that the grooved membranes with the higher surface area degrade significantly more methyl parathion than the non-grooved fibers. Controls of grooved and non-grooved fibrous membranes that were not grafted with the POM and exposed to methyl parathion for 24 h and 1 week showed no degradation. Using the equation derived from the calibration curve of methyl parathion, it was determined that the non-grooved fibrous membranes grafted with POMs resulted in a 17% decrease in the concentration of methyl parathion present in the methyl parathion/hexane solution after 24 h while the grooved cellulose acetate fibrous membranes grafted with POMs resulted in a 25% decrease in the concentration of methyl parathion present in the methyl parathion/hexane solution after 24 h. In terms of the amount of methyl parathion degraded by the POM, this translates to a decrease from 0.32 mg of methyl parathion in hexane solution to an amount of 0.26 mg of methyl parathion in the hexane solution as a result of POM grafted non-grooved fibers and 0.24 mg of methyl parathion in the hexane solution as a result of POM grafted grooved fibers after 24 h.
 | ||
| Fig. 8 Degradation of methyl parathion by POM grafted non-grooved 60/40 CA:PEO fibrous membranes and by grafted grooved cellulose acetate fibrous membranes after 1 week. | ||
After 1 week (168 h), differences in the degradation performance between grooved and non-grooved fibers became more distinct. It was determined that the non-grooved fibers grafted with POMs resulted in an 89% decrease in the concentration of methyl parathion present in the methyl parathion/hexane solution after 168 h while the grooved fibers grafted with POMs resulted in a 100% decrease in the concentration of methyl parathion present in the methyl parathion/hexane solution after only 5 days (120 h) as shown in Fig. 9 and 10. In terms of the amount of methyl parathion degraded by the POM, this translates to a decrease from 0.32 mg of methyl parathion in hexane solution to an amount of 0.035 mg of methyl parathion in the hexane solution as a result of POM grafted non-grooved fibers after 7 days and 0 mg of methyl parathion in the hexane solution as a result of POM grafted grooved fibers after 5 days. Therefore, the grooved fibers achieved a greater amount of methyl parathion degradation than the non-grooved fibers in a shorter amount of time. This difference in performance between electrospun fibers of the same diameter and cross-sectional shape can be attributed to the rougher surface morphology of the grooved fibers. Statistical analysis showed that the amount of degradation of methyl parathion by the grooved membranes was statistically significant with a p-value of 0.001. These results confirm the hypotheses that increasing the surface area of the substrate leads to an increase in POM binding sites and thus results in improved decontamination performance.
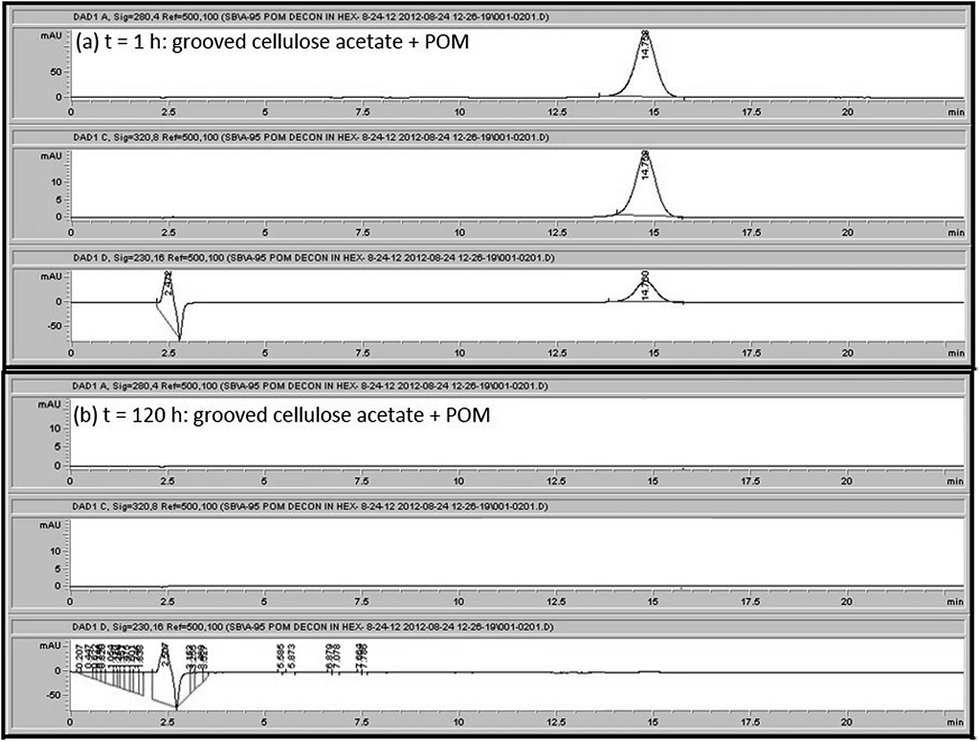 | ||
| Fig. 9 The HPLC measured decrease in methyl parathion peak area (a) after 1 h (b) and after 120 h by POM grafted grooved cellulose acetate membranes. | ||
 | ||
| Fig. 10 Degradation of methyl parathion by all POM grafted substrates after 1 week. | ||
The degradation results of methyl parathion by functionalized grooved cellulose membranes were comparable to the performance of the grooved cellulose acetate membranes achieving complete toxin degradation after five days. The fiber diameter of the grooved cellulose fibers was between 1 and 2 microns and thus the same as the grooved cellulose acetate fibers and the non-grooved counterparts, thereby illustrating that the grooved morphology of the fiber surface was the greater impact factor for increasing the methyl parathion degradation performance.
The degradation of methyl parathion by the functionalized cotton fabric and Tencel® fabric was statistically insignificant (p > 0.5) from each other, but slightly greater than that by the non-grooved fibers. Although the fiber diameters of the cotton and Tencel® substrates (both between 10 and 20 microns) were much greater than the fiber diameters of the electrospun membranes, the surface areas of the cotton and Tencel® fabric were not so much greater than that of the non-grooved membrane. Nevertheless, Cotton and Tencel® achieved higher percent POM loadings than the non-grooved membranes due to their complete cellulosic composition with more reaction sites for bound POM than the non-grooved cellulose acetate/polyethylene oxide blend. These results suggest that chemical composition yielding the most available POM binding sites in combination with the high surface area grooved fiber morphology enables the largest amount of POM particle retention per gram of fabric and thus the greatest amount of methyl parathion degradation.
In summary, a higher surface area substrate such as the grooved cellulose acetate fibers or the grooved cellulose degraded more methyl parathion than a lower surface area substrate such as the non-grooved 60/40 CA:PEO fibers, cotton, or Tencel®.
Regarding the degradation of methyl parathion and the expected degradation products of p-nitrophenol and methyl paraoxon, no peaks for degradation products were apparent in the HPLC-DAD results. For each degradation test, the standard solution of p-nitrophenol in water showed a peak at 320 nm with a 5 min retention time, and the standard solution of methyl paraoxon in hexane showed a peak at 280 nm at a 5 min retention time. The standard solution of methyl parathion showed a peak at 280 nm with a retention time of 15 min. Over the time of degradation testing, the peak area of the methyl parathion decreased at the 15 min retention time; however, no peaks were apparent at 280 nm or 320 nm with 5 min retention times indicating that the expected degradation products were not present as the POM degraded methyl parathion over time. It is possible that methyl parathion was degraded via hydrolysis. Degradation of methyl parathion by hydrolysis would have also produced the degradation product of p-nitrophenol and a second degradation product, diethylthiophosphate.33–35 Further investigation of the degradation mechanism(s) will be completed in future work.
Conclusions
The POM H5PV2Mo10O40 was synthesized although the precise structure is not known. The POM was used to functionalize fibers to obtain self-decontamination. The electrospinning of a 60/40 CA:PEO co-continuous fibrous membrane created non-grooved fiber morphology. By extracting the PEO from the membranes, a grooved fiber morphology was created to significantly increase the fiber surface area, which in turn provided greater fiber surface area for POM binding. Consequently, POM grafted grooved nanofibrous membranes exhibited approximately 25% more methyl parathion degradation in comparison with that of POM grafted non-grooved nanofibrous membranes. Our study also showed that the POM grafted grooved nanofibrous membranes outperformed conventional cotton or commercial Tencel® peachskin fabric grafted with POM.
The novel POM grafted grooved nanofibrous membranes have great potential in chemical protective apparel applications in defense or agriculture due to their enhanced self-decontamination of organophosphates and inherent porous structure for breathability. Further work is needed to examine thermal comfort and mechanical properties of such materials before they can be optimized for end-use products.
Acknowledgements
This research was funded by grants from the Cornell Agricultural Experiment Station, the North Central Regional Research Project NC 170 federal formula funds, Project NYC329407 received from the Cooperative State Research, Education, and Extension Service, the U.S. Department of Agriculture; the College of Human Ecology; and the American Association of Textile Chemists and Colorists. This work was supported in part by the Cornell Center for Materials Research with funding from the NSF MRSEC program (DMR-1120296). The authors appreciate helpful discussion with Professor Geoffrey Coates in the Department of Chemistry and Chemical Biology.Notes and references
- R. R. Ozer and J. L. Ferry, J. Phys. Chem. B, 2000, 104, 9444–9448 CrossRef CAS.
- L. Xu, E. Boring and C. L. Hill, J. Catal., 2000, 195, 394–405 CrossRef CAS.
- J. Walker, H. Schreuder-Gibson, W. Yeomans, D. Ball, F. Hoskin and C. Hill, Development of Self-Detoxifying Materials for Chemical Protective Clothing, Army Natick Soldier Center, MA, 2003 Search PubMed.
- R. D. Gall, C. L. Hill and J. E. Walker, Chem. Mater., 1996, 8, 2523–2527 CrossRef CAS.
- E. A. Boring, C. L. Hill, J. T. Rhule and L. Xu, WO2001034279 A3, 2002.
- U. Drechsler, W. Singh and A. Sharma, US 2009/0012204 A1, 2009.
- L. E. Lange and S. K. Obendorf, ACS Appl. Mater. Interfaces, 2015, 7, 3974–3980 CAS.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed.
- G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437–441 CrossRef CAS.
- J. T. McCann, D. Li and Y. Xia, J. Mater. Chem., 2005, 15, 735 RSC.
- C. Tang, P. Chen and H. Liu, Polym. Eng. Sci., 2008, 48, 1296–1303 CAS.
- W. Zhang, D. Yao, Q. Zhang, J. G. Zhou and P. I. Lelkes, Biofabrication, 2010, 2, 35006 CrossRef PubMed.
- D. J. Woo and S. K. Obendorf, RSC Adv., 2014, 4, 15727–15735 RSC.
- H. Liu and Y.-L. Hsieh, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2119–2129 CrossRef CAS.
- V. Dixit, J. Tewari and S. K. Obendorf, Arch. Environ. Contam. Toxicol., 2010, 59, 417–423 CrossRef CAS PubMed.
- K. Tan and S. K. Obendorf, J. Membr. Sci., 2006, 274, 150–158 CrossRef CAS.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, 1983 Search PubMed.
- M. Misono and T. Okuhara, CHEMTECH, 1993, 23, 11 Search PubMed.
- L. Pettersson, I. Andersson, J. H. Grate and A. Selling, Inorg. Chem., 1994, 33, 982–993 CrossRef CAS.
- A. Selling, I. Andersson, L. Pettersson, C. M. Schramm, S. L. Downey and J. H. Grate, Inorg. Chem., 1994, 33, 3141–3150 CrossRef CAS.
- U. G. Nielsen, A. Hazell, J. Skibsted, H. J. Jakobsen and C. J. McKenzie, CrystEngComm, 2010, 12, 2826–2834 RSC.
- J. Arichi, M. M. Pereira, P. M. Esteves and B. Louis, Solid State Sci., 2010, 12, 1866–1869 CrossRef CAS.
- J. Arichi, M. Eternot and B. Louis, Catal. Today, 2008, 138, 117–122 CrossRef CAS.
- R. A. Nyquist and R. O. Kagel, Infrared spectra of inorganic compounds (3800-45cm... [to the minus one]), Academic Press, New York, London (24 Oval Rd, N.W.1), 1971 Search PubMed.
- J. W. Robinson, Practical handbook of spectroscopy, CRC Press, Boca Raton, 1991 Search PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- M. D. Zidan and A. W. Allaf, Spectrochim. Acta, Part A, 2000, 56, 2693–2698 CrossRef CAS.
- A. Popa, V. Sasca, M. Stefanescu, E. Kis and R. Marinkovic-Neducin, J. Serb. Chem. Soc., 2006, 71, 235–249 CrossRef CAS.
- L. M. Gratton, S. Paglia, F. Scattaglia and M. Cavallini, Appl. Spectrosc., 1978, 32, 310–316 CrossRef CAS.
- P. Larkin, Infrared and Raman Spectroscopy; Principles and Spectral Interpretation, Elsevier, 2011 Search PubMed.
- H. S. Blair and M. J. McElroy, J. Appl. Polym. Sci., 1976, 20, 2955–2961 CrossRef CAS.
- R. Neumann, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1997, pp. 317–370 Search PubMed.
- D. C. Fee, D. R. Gard and C.-H. Yang, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000 Search PubMed.
- R. L. Metcalf, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- J. R. Morrow and W. C. Trogler, Inorg. Chem., 1989, 28, 2330–2333 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04456e |
| ‡ Polyoxometalate (POM). |
| § 10-Molybdo-2-vanadophosphoric acid (H5PV2Mo10O40). |
| ¶ Chemical warfare agent (CWA). |
| || VX is an abbreviation for nerve gas used as a chemical warfare agent. |
| ** 2-Chloroethyl ethyl sulfide (CEES) is a sulfur mustard analog. Sulfur mustard is used as a chemical warfare agent. |
| †† Hexamethylene diisocyanate (HMDI). |
| ‡‡ Fourier transform infrared spectroscopy (FTIR). |
| §§ Room temperature (RT). |
| ¶¶ Thermogravimetric analysis (TGA). |
| |||| Scanning Electron Microscopy (SEM). |
| *** High performance liquid chromatography/diode-array UV/VIS detector (HPLC-DAD). |
| ††† Analysis of Varianace (ANOVA) using JMP 9.0 statistical software. |
| ‡‡‡ ImageJ is an image processing program. |
| This journal is © The Royal Society of Chemistry 2016 |