
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advanced oxidation processes for the removal of [bmim][Sal] third generation ionic liquids: effect of water matrices and intermediates identification†
Nemanja
Banić
,
Biljana
Abramović
*,
Filip
Šibul
,
Dejan
Orčić
,
Malcolm
Watson
,
Milan
Vraneš
and
Slobodan
Gadžurić
University of Novi Sad, Faculty of Sciences, Department of Chemistry, Biochemistry and Environmental Protection, Trg D. Obradovića 3, 21000 Novi Sad, Serbia. E-mail: biljana.abramovic@dh.uns.ac.rs; Fax: +381 21 454065; Tel: +381 21 4852753
First published on 23rd May 2016
Abstract
Unique properties of ionic liquids make them green alternatives for conventional volatile organic compounds. Due to increased production and the high stability of these substances, they could be classified as persistent pollutants and could break through classical treatment systems into natural waters. A preliminary ionic liquid hydrolysis study demonstrated a pH dependent degradation profile with a significant decrease in hydrolysis efficiency as pH lowered from 10.0 to 2.8. In order to examine future prospects for ionic liquid removal, different advanced oxidation processes (TiO2 Degussa P25/H2O2, TiO2 Degussa P25, 7.2Fe/TiO2/H2O2, and H2O2) were studied for their applicability in the degradation of imidazolium-based ionic liquids in aqueous solution. These processes were conducted in the dark as well as in the presence of UVA and simulated sunlight (SS) radiation. Among the investigated dark processes, the 7.2Fe/TiO2/H2O2 system showed the highest efficiency, which can be attributed to a dark heterogeneous Fenton process. Otherwise, the most efficient among all the studied degradation processes was the UVA/TiO2 Degussa P25/H2O2 process. In order to make degradation processes more similar to that of the practical process SS radiation was used. Among studied processes, the 7.2Fe/TiO2/H2O2 system showed the greatest potential for the removal of ionic liquids. Also, it was observed that the impact of anions on the cation degradation efficiency was much more pronounced. Due to the possible fate of ionic liquids in the environment, for five different waters (pond, rain, tap, river, and condensate) degradations in the dark and under simulated sunlight were studied. For all processes, and all water types in the presence of SS radiation a remarkable positive effect of naturally dissolved organic matter on the degradation efficiency was observed. Also, in all experiments, the anion was less stable than the cation. The major photodegradation products identified using liquid chromatography-mass spectrometry (HPLC-MS/MS) techniques were hydroxylated compounds.
1. Introduction
Room-temperature ionic liquids are receiving considerable attention as green, high-tech reaction media of the future.1 This can be explained by the fact that ionic liquids possess negligible vapor pressure, so that they can be a good alternative to the emissions of toxic vapors from conventional molecular organic solvents.2 In addition to these features, ionic liquids have other good properties, such as good thermal stability, a wide electrochemical potential window, high electric conductivity and miscibility with water or organic solvents which make them interesting for application in different fields.3,4 Up to now the ionic liquids have been used in lithium ion batteries,5 gas separation, compression and handling,6,7 metal plating,5,8,9 dyesensitized solar cells, and as paint or polymer additives.10,11Furthermore, the third generation of ionic liquids includes salts with a melting point below 100 °C that contain biologically active cations and/or anions in the structure.12,13 A special attention of the scientific community in the recent years has been paid to ionic liquids with salicylate anion, since they show possibility of a wide range of applications in the field of pharmaceutical industry,14 the synthesis of nanoparticles,15 heat storage,16 electrodeposition of transition metals17etc. Complexing (chelating) ability of salicylate anions was used for the synthesis of the hydrophobic ionic liquids for selective metal (metalloid) extraction from the industrial and the communal wastewaters.18–21
On the other hand a broad implementation of ionic liquids leads us to the following question: what will be the outcome when these substances get through the environment? It can be assumed that their low vapor pressure provides air protection from the emission of toxic vapors, but on the other hand their stability makes them hazardous to water and soil.22 Due to its high stability in the near future it can be expected the presence of significant amounts of ionic liquids in technological wastewaters and their break through classical treatment systems into natural waters.1 Therefore, information concerning their fate in the aquatic environment is very important.
Also, some studies showed that ionic liquids are more toxic to the cells in comparison to conventional solvents.1 These studies are in good agreement with other studies in which the most common imidazolium based compounds showed high resistance toward microbial degradation.22
It can be also assumed that among the various transformation processes (biotic and abiotic), photostability is an important factor influencing the fate of ionic liquids in the environment. Thus, it is very important to find an efficient method for a complete and safe removal of ionic liquids from the environment.23–25 The advanced oxidation processes (AOPs), for example, UVA/H2O2, UVA/Fe2+/H2O2, and UVA/TiO2 are potentially useful for wastewater treating because they generate hydroxyl radicals,26–28 a powerful nonspecific oxidant, and can provide an almost total degradation.29 Among numerous AOPs, Fenton's process is one of the promising technologies because of the high oxidation potential and relatively low cost.30 This process can be enhanced by visible light due to the decomposition of the photo-active Fe(OH)2+ species producing additional OH-radicals in solution.31 In these processes participation of iron oxide minerals (hematite α-Fe2O3, goethite α-FeOOH, magnetite Fe3O4 and ferrihydrite) as the catalysts in the presence of hydrogen peroxide was also effective in the past, because uniform rates of hydroxyl radical generation at high concentration of hydrogen peroxide used. Moreover α-FeOOH/H2O2 system has a number of advantages as compared to a homogenous Fenton's system with respect to reaction pH range and the removal of iron.32
In this study efficiency of ionic liquid degradation was investigated and compared by different AOPs, including direct and H2O2 indirect photolysis, as well as heterogeneous photocatalysis using TiO2 and 7.2Fe/TiO2 catalysts in the presence of UVA/simulated sunlight (SS) radiation. In contrast to previously studied ionic liquids that contain an inorganic anion i.e. chloride1,22,33–35 or bromide,36 in this paper an ionic liquid in which both components were organic ions was investigated. Also, mutual influence of anion and cation was studied in detail. For broaden the application of used AOPs, the photocatalytic degradation of different kind of ionic liquids were also studied. Because there is no information in the available literature on the photodegradation of ionic liquid under natural aquatic conditions, the present study deals with the influence of water matrices on the ionic liquid photodegradation efficiency. In this study five different types of waters was used HPLC-MS/MS technique was used to track the 23 intermediate products formed during the course of ionic liquid degradation.
2. Experimental section
2.1 Chemicals and solutions
All chemicals were of reagent grade and were used without purification. Studied 1-butyl-3-methylimidazolium salicylate ([bmim][Sal]) ionic liquid was synthesized according to the procedure mentioned in the literature.17 The ionic liquid 1-benzyl-3-methylimidazolium chloride ([Bzmim]Cl) was manufactured by Iolitec-Ionic Liquids Technologies GmbH, Heilbronn (Germany) and 1-butyl-3-methylimidazolium chloride ([bmim]Cl) by Merck. Other ionic liquids selected for these studies were obtained from the ionic liquids collection of the working group of Dr Milan Vraneš. They were tetrabutylammonium salicylate ([TBA][Sal]), 1-(3-hydroxypropyl)-3-methylimidazolium salicylate ([Hpmim][Sal]) and imidazolium salicylate ([im][Sal]). Sodium salicylate ([Sal]Na) was produced by Merck. 30% H2O2, was obtained from Centrohem (Stara Pazova, Serbia); 99.8% acetonitrile (J. T. Baker); 85% H3PO4 (Lachema, Neratovice, Czech Republic); H2SO4, formic acid, ammonium acetate (Merck); and NaOH (ZorkaPharm, Šabac, Serbia). All solutions were prepared using doubly distilled water. The concentration of ([bmim][Sal]) stock solution used in photodegradation experiments was 0.38 mmol L−1. Photocatalyst with 7.24% of Fe (w/w, denoted as 7.2Fe/TiO2) synthesized by a simple deposition–precipitation method,27 TiO2 support obtained from Molar Chemicals KFT (Hungary) and TiO2 P25 manufactured by Evonik Corporation (USA) (75% anatase and 25% rutile form, surface area of 50 m2 g−1 according to the manufacturer's specification, with average particle size about 20 nm,37) were used as photocatalysts.2.2 Degradation experiments
All experiments were carried out in a batch reactor made of Pyrex glass (total volume of cca 100 mL, solution depth 46 mm). The UVA radiation was provided from a 125 W high-pressure mercury lamp (λ > 290 nm, Philips HPL-N, Netherlands, emission bands in UV region at 304, 314, 335, and 366 nm, with the maximum emission at 366 nm). Intensity of UVA radiation was 3.57 × 10−3 W cm−2. Irradiation with SS light was performed using a set of four 35 W halogen lamps (λ > 300 nm, VITO, MR 16, China), placed in a symmetrical way around the reactor. UVA and vis radiation intensities for applied SS light were 1.8 × 10−4 W cm−2 and 116.2 × 10−3 W cm−2, respectively.A solution of 30 mL [bmim][Sal] containing 1.67 g L−1 of catalyst and/or 0.45 mmol L−1 H2O2 with successive addition of H2O2 (70 μL each hour during the degradation) was used (except for the study of hydrolysis and direct photolysis). All experiments were performed at pH 2.8 (except in the case of hydrolysis study). A value of 2.8 was chosen because it was optimal value for 7.2Fe/TiO2/H2O2 system.27 The H2SO4 and NaOH solutions were used to adjust the pH of reaction solutions. The lamps were turned on for 15 min prior to degradation to obtain a constant light intensity output. At the same time reaction solution was stirred continuously in the dark in order to ensure equilibrium adsorption of [bmim][Sal] on the photocatalyst.
To ensure a constant O2 the reaction solution was continuously purged to the bottom of the reactor with molecular oxygen (99.99% purity) at a constant flow rate of 5 mL min−1. Furthermore the solution was homogenized with the aid of a stirring bar, to ensure completely mixed batch conditions. The temperature of the reaction solution was kept at 25 ± 0.5 °C throughout the experiment by water circulation system. Dark experiments were conducted under identical conditions but without radiation.
The matrix effects of five water types on the efficiency of ionic liquid degradation were evaluated. For this purpose we used the following samples: river water, rain water, pond water, tap water, and condensate from air conditioner. The river water sample was collected from the Danube (Novi Sad, Serbia) in July 2014. Of the same year in the urban area of Novi Sad, rain water and pond water samples were collected during the rainy periods between the ninth and fourteenth of July. In the same month tap water sample was collected from public drinking water supply system of Bački Jarak (Serbia) while the condensate water from the air conditioner harvested in one flat that is also located at the territory of Novi Sad. To avoid microbial growth, water samples were sterilized by filtration and all glassware was sterilized by autoclaving for 30 min at 140 °C.
2.3 Analytical methods
For the kinetic studies of ionic liquid removal, 0.5 mL of the reaction mixture were sampled at the beginning of the experiment and at regular time intervals, followed by filtration through Millipore (Millex-GV, 0.22 μm) membrane filter (in the presence of catalyst). The lack of ionic liquid adsorption onto the filters was confirmed by a preliminary test. The HPLC-DAD analyses were performed on a Shimadzu 20A series ultra-fast liquid chromatograph (UFLC, Shimadzu Cooperation, Kyoto, Japan), equipped with UV-vis diode array detection and polar CN column (Inertsil CN-3 column; 150 mm × 4.6 mm i.d., particle size 3 μm). The injection volume was 20 μL. Column temperature was held at 25 °C, mobile phase was a mixture of 0.1% H3PO4–ACN (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v/v), pH 2.56, and the total flow rate was 0.8 mL min−1. Ionic liquid elution was monitored at 210 nm. The retention time of ionic liquid's cation was 1.44 min while the same for the anion was 6.58 min.
:4, v/v), pH 2.56, and the total flow rate was 0.8 mL min−1. Ionic liquid elution was monitored at 210 nm. The retention time of ionic liquid's cation was 1.44 min while the same for the anion was 6.58 min.
For collected water samples physicochemical characteristics are given in Table 1.
Dissolved organic carbon (DOC) of different water samples (filtered through 0.22 micrometer filter) was determined on an Elementar Liqui TOC II (Germany) in accordance with Standard US EPA Method 9060A. Before measurements samples were acidified.
The radiation fluxes were measured using a Delta Ohm HD 2102.2 (Padova, Italy) radiometer which was fitted with the LP 471 UVA (spectral range 315–400 nm) and LP 471 RAD (spectral range 400–1050 nm) sensors.
The conductivity measurements were performed using a conductivity meter type Cond 3210 (WTW, Weilheim, Germany) equipped with a standard conductivity measuring cell TetraCon 325.
The pH measurements were made using a Hanna Instruments combined glass electrode (Kehl, Gemany), on a previously calibrated pH-meter (Iskra, Kranj, SFRJ).
In addition, in order to gain insight into the relationship between the elements present in the different water matrices and the efficiency of ionic liquid degradation, the signal intensities obtained by scanning the waters by inductively coupled plasma mass spectrometry (ICP-MS) are given in Table S1, ESI.† The ICP-MS analysis was carried out on an Agilent 7700e ICP-MS (Hachioji, Japan), with a collision cell operated in helium mode. General purpose plasma conditions were applied, with a power of 1550 W and a sampling depth of 10 mm. A full mass scan was carried out, from Li 7 to Pb 208, and the resulting spectra analysed using the Agilent MassHunter software.
For the HPLC-ESI-MS/MS identification of intermediates, 0.38 mmol L−1 of ionic liquid solution was prepared, and UVA/TiO2 P25/H2O2 system was used. Filtration was carried out to separate the photocatalyst particles as previously described. Then, a 20 μL of sample was injected and analyzed by an Agilent Technologies 1200 series HPLC with Agilent Technologies 6410A series electrospray ionization triple-quadrupole MS/MS, using the aforementioned polar CN column held at 25 °C. The mobile phase, consisting of 0.1% aqueous formic acid and ACN, was delivered at flow rate 0.8 mL min−1.
In negative mode, isocratic elution (20% ACN) was used. In positive mode, 100 mmol L−1 ammonium acetate was added to aqueous phase to enhance retention of charged quaternary nitrogen compounds (that otherwise elute at dead time), and gradient mode (0 min 20% ACN, 4 min 20% ACN, 6 min 100%, post time 3 min) was employed to facilitate separation. Analytes were ionized using the electrospray ion source, with nitrogen as the drying gas (temperature 350 °C, flow 10 L−1 min−1) and nebulizer gas (50 psi), and a capillary voltage of 4.0 kV. High purity nitrogen was used as the collision gas. Full scan mode (m/z range 50–500, scan time 100 ms, fragmentor voltage 100 V), using both polarities, was used to detect degradation intermediates, determine their molecular weight and select precursor ions for MS2 experiments. To obtain structural information and identify the detected compounds, product ion scan analysis (MS2 experiment) was used, with [M + H]+ and [M − H]− ions as precursors, and collision energies in 10–40 V interval (10 V increments).
3. Results and discussion
3.1 Effect of pH on ionic liquid hydrolysis
The degradation of ionic liquid in aqueous solution in the absence of light at ambient temperature of 25 °C was monitored at different pH values. As can be seen from Fig. 1a after 93 days in pH range between pH 2.8 and 10.0, [bmim]+ (i.e. cation of the ionic liquid) was stable to hydrolysis. Unlike the [bmim]+, [Sal]− (i.e. anion of the ionic liquid) was less stable as the pH increased (Fig. 1b). Namely, at pH 7.0 and 10.0 after 51 days 41.5, and 100% of the [Sal]− was hydrolyzed, respectively. Also, at pH 2.8 no noticeable hydrolysis of [Sal]− occurred. Finally, it can be concluded that the ionic liquid was stable at pH 2.8 toward hydrolysis. Therefore, all subsequent experiments were carried out at pH 2.8.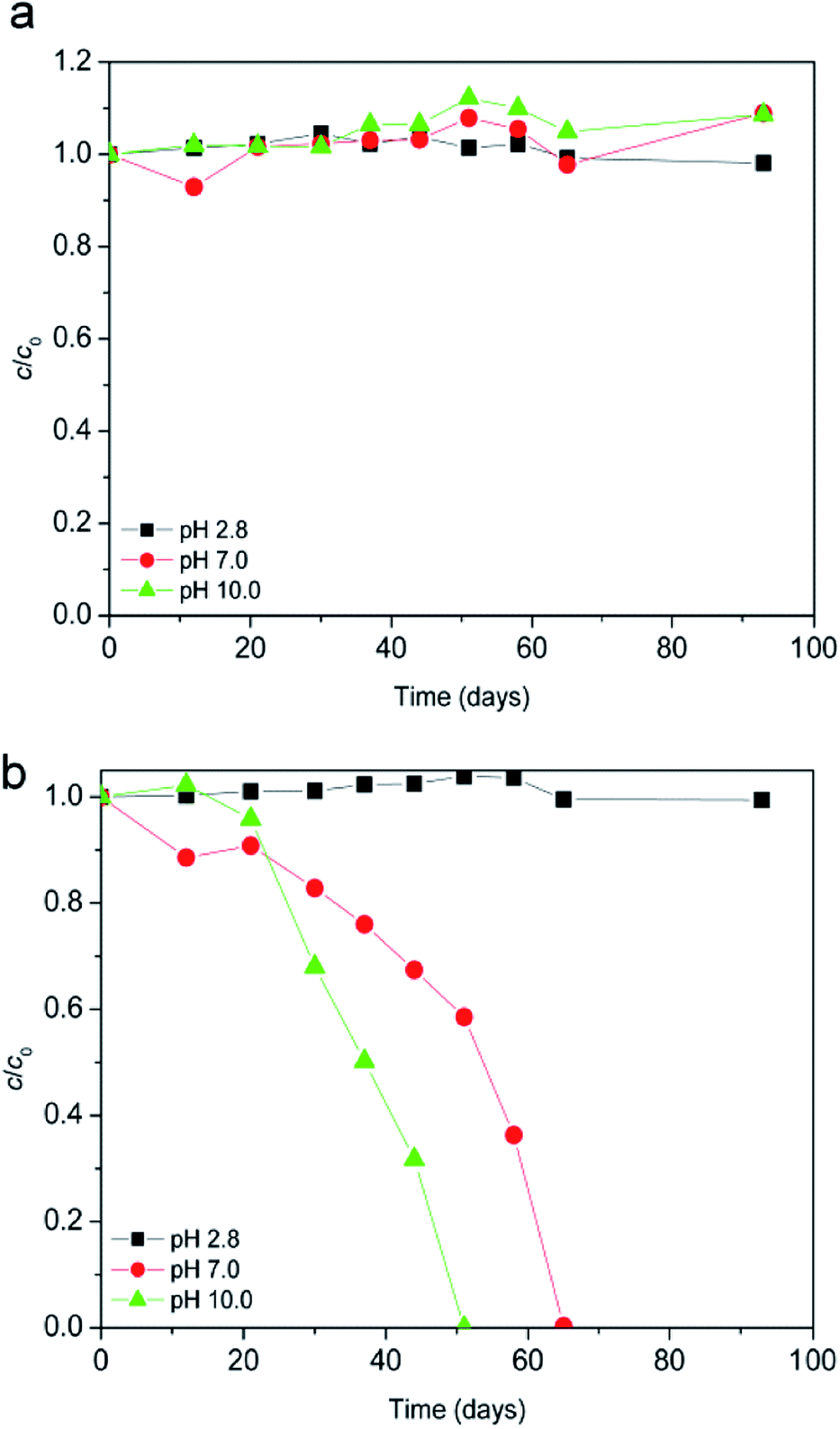 | ||
| Fig. 1 Effect of pH on ionic liquid (c0 = 0.38 mmol L−1) hydrolysis: (a) [bmim]+; (b) [Sal]−. | ||
Every degradation study was performed with both ions of ionic liquid (except where otherwise stated), but for the clarity reasons the results for [bmim]+ and [Sal]− were in each figure separately shown.
3.2 Preliminary dark experiments
Since the suspension was stirred continuously in the dark for 15 min prior to the degradation, it was examined whether this contact time was sufficient for [bmim][Sal] to reach adsorption–desorption equilibrium on the photocatalyst. Even after 180 min, the removal of [bmim][Sal] by dark adsorption at natural pH and pH 2.8 was almost negligible (Fig. 2).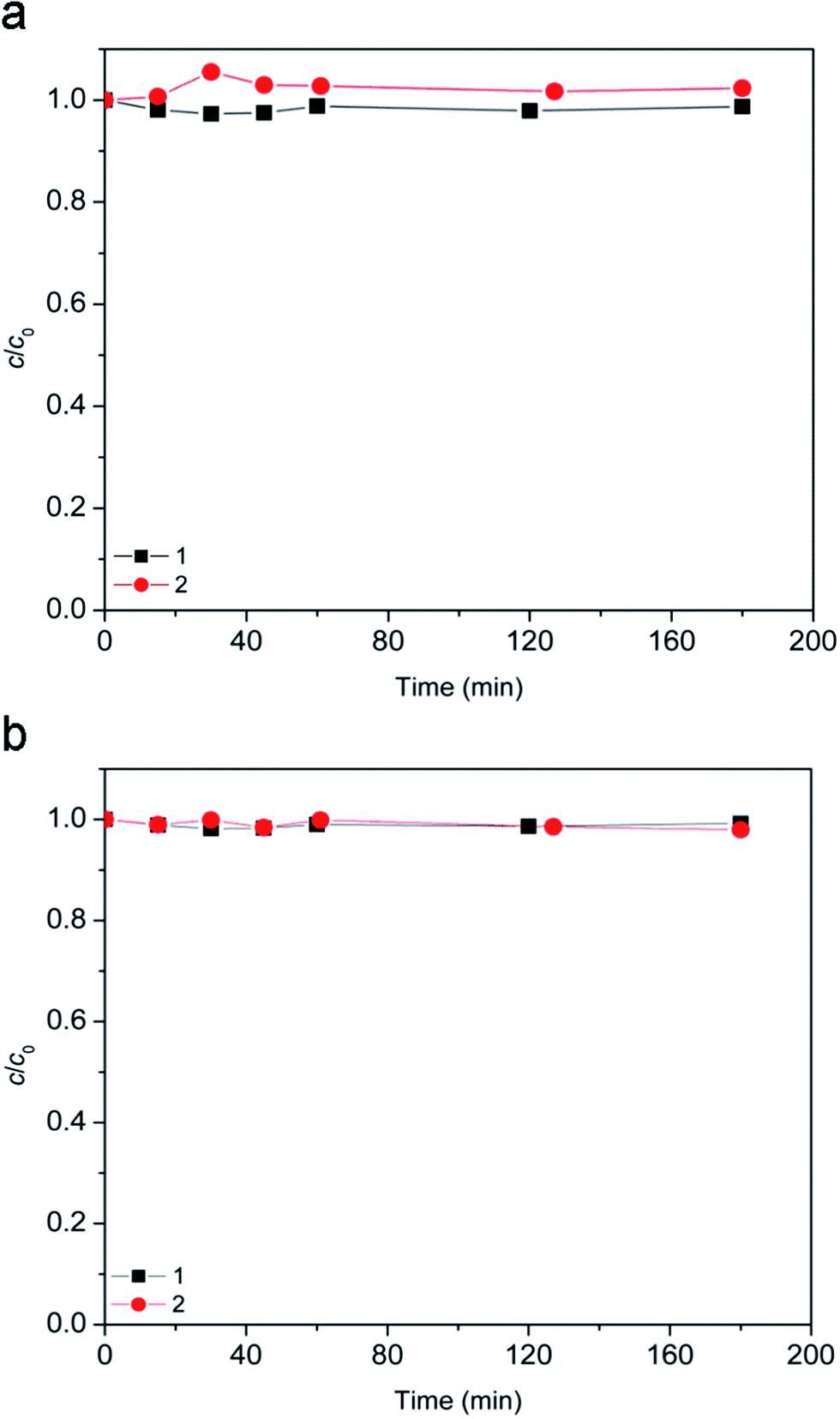 | ||
| Fig. 2 Evolution of ionic liquid (c0 = 0.38 mmol L−1) adsorption along the time: (a) [bmim]+ and (b) [Sal]−. Applied conditions: (1) 7.2Fe/TiO2 (1.67 g L−1) at natural pH; (2) 7.2Fe/TiO2 (1.67 g L−1) at pH 2.8. | ||
Furthermore, obtained results for the experiments conducted in dark showed that the ionic liquid was stable in the presence of H2O2 alone (Fig. 3, curve 1), as well as in a combination of the TiO2 photocatalysts (TiO2 P25 or TiO2 support) and H2O2 (Fig. 3, curves 3 and 4). Also, using 7.2Fe/TiO2 did not cause the ionic liquid degradation (Fig. 3, curve 5).
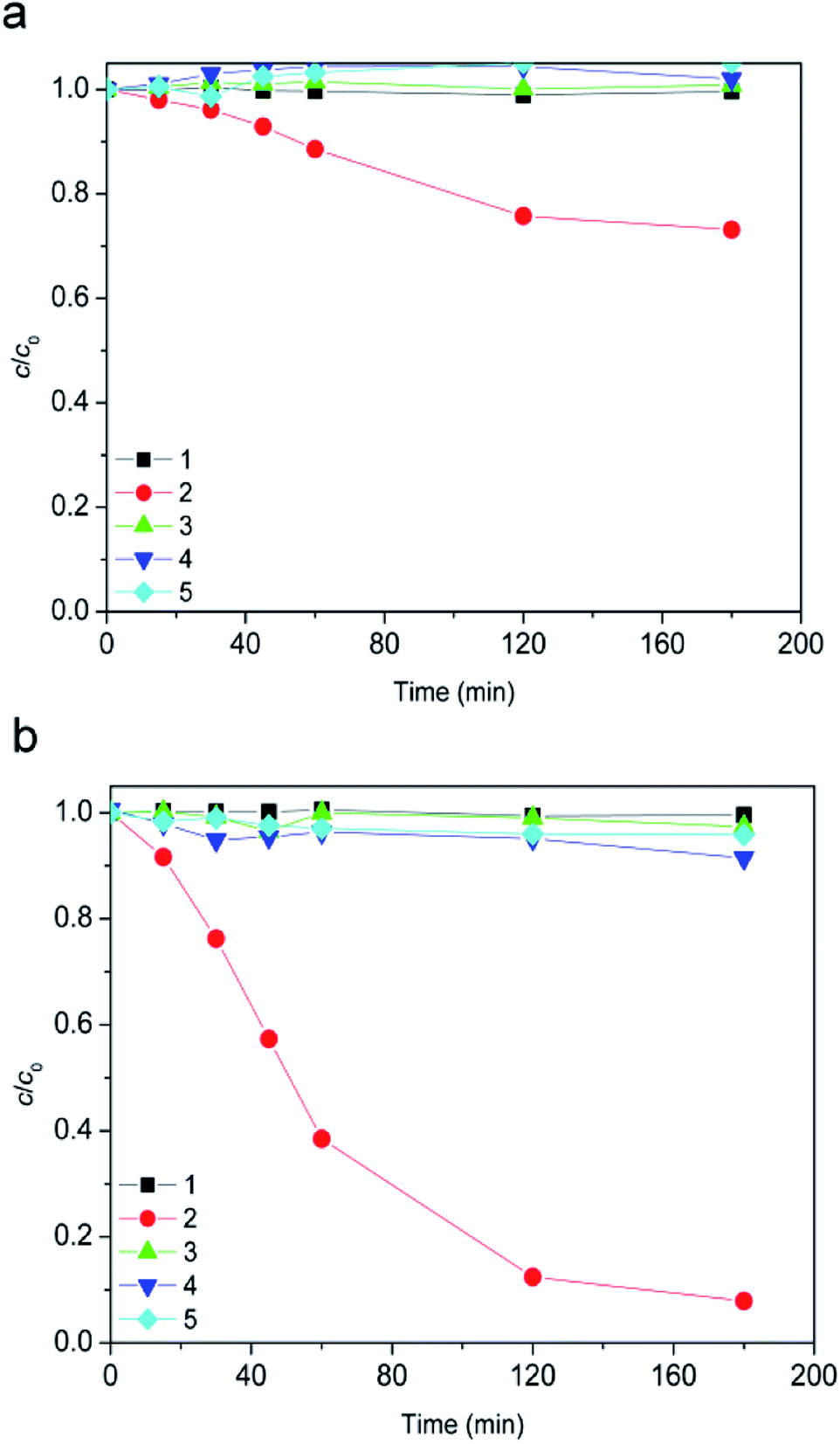 | ||
| Fig. 3 Evolution of ionic liquid (c0 = 0.38 mmol L−1) dark conversion along the time: (a) [bmim]+ and (b) [Sal]−. Reaction conditions: (1) 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (2) 7.2Fe/TiO2 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (3) TiO2 support (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (4) TiO2 P25 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (5) 7.2Fe/TiO2 (1.67 g L−1) at pH 2.8. | ||
However, in the presence of H2O2 and 7.2Fe/TiO2 after 180 minutes 27% of [bmim]+ (Fig. 3a, curve 2), and 92% of [Sal]− (Fig. 3b, curve 2) has been degraded. This behavior can be attributed to the Fenton process.
According to our previous examinations and since all of the experiments were conducted at pH 2.8, for 7.2Fe/TiO2/H2O2 system is expected that the concentration of iron in solution will not exceed 8 ppb. In compliance with this, the degradation efficiency was studied in the presence of 8 ppb dissolved iron(III) and appropriate amounts of hydrogen peroxide. After 180 min it has been found that the ionic liquid degradation efficiency at this level of dissolved iron concentration (homogeneous Fenton processes) was negligible.
The same conclusion was derived from the experiment with 60 ppb of Fe(III). This experimental evidence implies that the overall degradation efficiency originates from the heterogeneous Fenton process (reactions (1)–(5)).38
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeIII–OH + H2O2 ⇄ (H2O2)s FeIII–OH + H2O2 ⇄ (H2O2)s | (1) |
| (H2O2)s → FeII + H2O + HO2˙ | (2) |
| FeII + H2O2 → FeIII–OH + HO˙ | (3) |
| HO2˙ ⇄ H+ + O2˙− | (4) |
| FeIII–OH + HO2˙/O2˙− → FeII + H2O/OH− + O2 | (5) |
Finally, it can be concluded that within this process existence of surface bonded iron oxide was crucial. This statement is based on our previous experiments27 which were carried out with PrecFe (iron particles contained in 7.2Fe/TiO2 photocatalyst) in the presence of H2O2, and it was found that the degradation efficiency was practically the same to that in its absence. This was explained with the fact that the solution did not practically contain Fe3+ ions. However, the presence of H2O2 was increased the catalytic efficiency of 7.2Fe/TiO2 to a higher extent than of the TiO2 support and PrecFe, which suggests that H2O2 plays a different role, i.e., in the case of 7.2Fe/TiO2/H2O2, in addition to the electron-acceptor role, H2O2 is also involved in the heterogeneous photo-Fenton process.
3.3 UVA photodegradation studies
The [bmim]+ strongly absorbs UV radiation over a range of wavelengths, from 200 to 235 nm, with the maximum absorption band at 210 nm. On the other hand, the [Sal]− showed UV absorption at a broader range between 200 and 335 nm, with three absorption maxima at 205, 236, and the lowest at 303 nm. Since the UV radiation of used lamp below of closely 275 nm cannot pass through Pyrex glass,25 no direct photolysis of [bmim]+ could be expected. Also, observing the radiation of lamp which passed through the wall of the reactor and the absorption spectrum of absorbed radiation by [Sal]− in the range of 275 to 335 nm, a direct photolysis of the [Sal]− could be possible.After 180 minutes of radiation, no significant change in the [bmim]+ concentration could be observed (Fig. 4a, curve 1). Following the same radiation period 7% [Sal]− was degraded (Fig. 4b, curve 1).
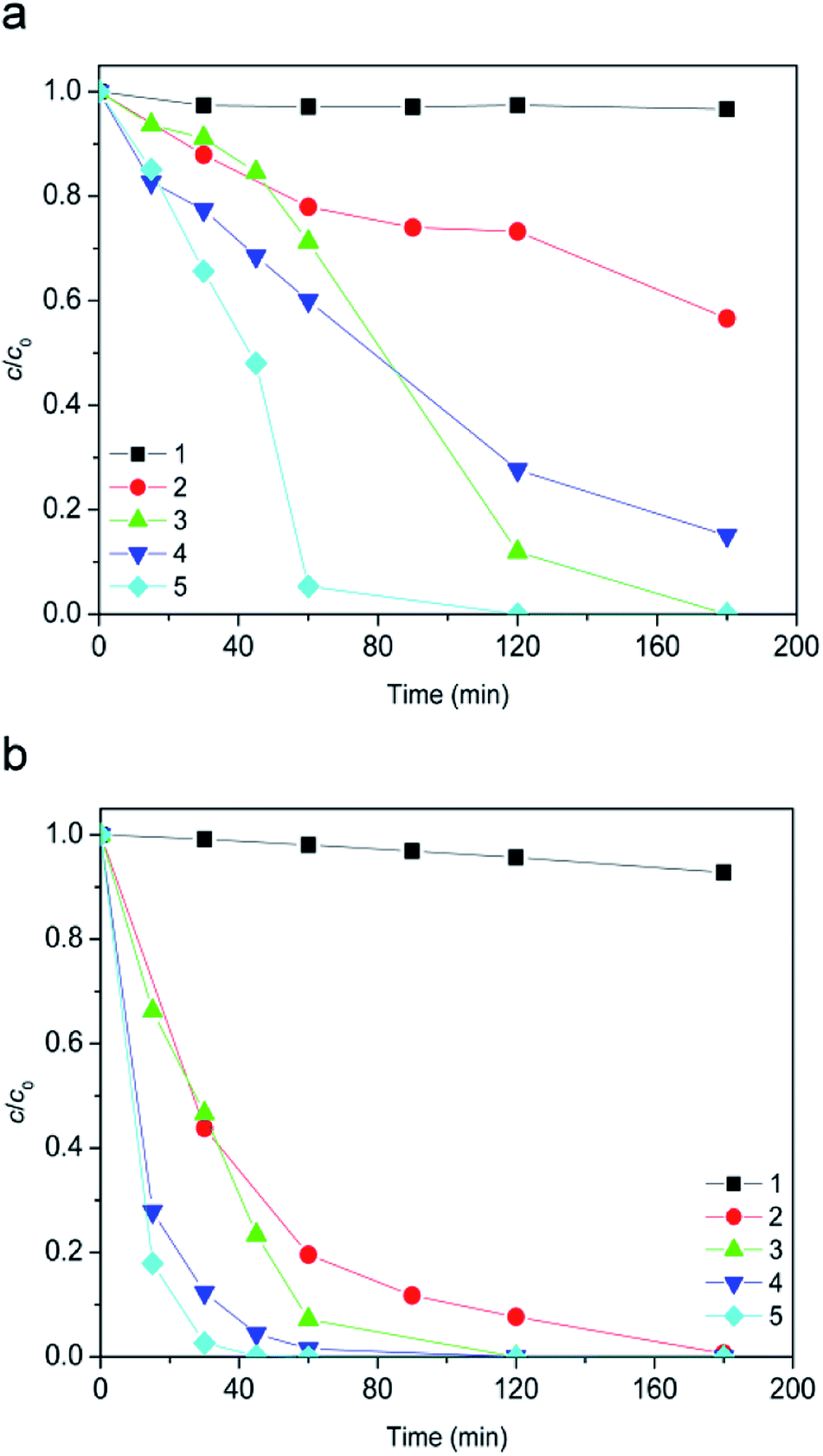 | ||
| Fig. 4 Evolution of ionic liquid (c0 = 0.38 mmol L−1) degradation along the time using UVA radiation: (a) [bmim]+ and (b) [Sal]−. Applied AOPs: (1) direct photolysis at pH 2.8; (2) 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (3) TiO2 P25 (1.67 g L−1); (4) 7.2Fe/TiO2 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (5) TiO2 P25 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2. | ||
Besides direct photolysis in Fig. 4 was presented the efficiency of other AOPs that can be potentially used for the ionic liquid degradation. This includes the results obtained using indirect photolysis by H2O2 (Fig. 4, curve 2), and photocatalysis with TiO2 P25 (with and without H2O2, Fig. 4, curves 3 and 5), as well as with 7.2Fe/TiO2/H2O2 system (Fig. 4, curve 4). In experiments in which H2O2 was employed, because of its depletion, H2O2 was added successively (70 μL each hour during the radiation). By comparing the results presented in Fig. 3 and 4 it can be noticed that the presence of UVA radiation for all investigated processes significantly increased efficiency of ionic liquid degradation. The contribution of UVA radiation on the studied [bmim]+ degradation decreases in the following order: UVA/TiO2 P25/H2O2 > UVA/TiO2 P25 > UVA/7.2Fe/TiO2/H2O2 > UVA/H2O2 > UVA. For [Sal]− the above-mentioned contribution sequence would be as follows: UVA/TiO2 P25/H2O2 > UVA/TiO2 P25 > UVA/H2O2 > UVA/7.2Fe/TiO2/H2O2 > UVA.
The most efficient degradation process included H2O2 coupled with catalytic degradation and the complete removal of [bmim]+ and [Sal]− required only 120, and 45 min, respectively. Somewhat lowered, but still high efficiency of TiO2 P25 photocatalysis in the absence of H2O2 resulting from the absorption spectrum of TiO2 P25.39 After 180 min of radiation, increased degradation efficiency for heterogeneous 7.2Fe/TiO2/H2O2 Fenton system which comes from UVA radiation (heterogeneous photo-Fenton) was 58.1% in the case of [bmim]+, and only 7.3% in the case of the [Sal]−.
Also increased efficiency of indirect photolysis can be explained by the fact that radiation with wavelengths less than 300 nm can cause photolysis of hydrogen peroxide.39
3.4 Simulated sunlight photodegradation studies
Further tests were conducted to examine the performance of different AOPs through degradation of ionic liquid under the SS radiation for the first time. Using this kind of the light source, the degradation process was more similar to that of the practical process which proceeds under sunlight.Comparing obtained results under SS radiation (Fig. 5) with those obtained for the same processes which were carried out in the dark (Fig. 3), in all cases increasing efficiency can be observed.
 | ||
| Fig. 5 Evolution of ionic liquid (c0 = 0.38 mmol L−1) degradation along the time using SS radiation: (a) [bmim]+ and (b) [Sal]−. Applied AOPs: (1) 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8; (2) TiO2 P25 (1.67 g L−1); (3) 7.2Fe/TiO2 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8. | ||
For the studied SS/H2O2, SS/TiO2 P25 and SS/7.2Fe/TiO2/H2O2 processes, these increases of efficiency were different for cation and anion. Namely, for the [bmim]+ (Fig. 5a) were 17%, 9%, and 4.5%, respectively, while the same in the case of [Sal]− (Fig. 5b) were 74%, 52%, and 5.3%, respectively. In this case, the spectral energy distribution of used SS source was more suited for conducting indirect photolysis in relation to the photocatalysis. On the other hand, for the SS/7.2Fe/TiO2/H2O2 process it was noticed that the contribution of the heterogeneous photo-Fenton oxidation was negligible compared to the dark heterogeneous Fenton oxidation.
Also, if compared Fig. 5 and 4, for all of the studied processes, ionic liquid photodegradation efficiency was significantly lower using SS radiation, compared to that in the presence of UVA radiation.
Further research has been focused on studying mutual facilitation of [bmim]+ and [Sal]− on the photodegradation efficiency in the presence of the SS radiation. For this purpose, the SS/TiO2 P25 process was chosen. By applying this process the degradation efficiency in the presence of only [bmim]+ or only [Sal]− has been studied (Fig. 6) and compared with those obtained when they are in a mixture (referring to the ionic liquid, Fig. 5). When the degradation efficiency of individual ions was studied, [bmim]+ was present in the form of chlorides, while the [Sal]− was present in the form of the sodium salt. Comparing results presented in Fig. 5 and 6 it can be seen that after 180 min of only [Sal]− degradation, efficiency decreased by 5.4%, while in the case of [bmim]+, efficiency was increased for 36%. Based on previous conclusions it can be established that the impact of [Sal]− on the [bmim]+ degradation efficiency was much more pronounced. It was also noted that in all experiments when both ions are present in the mixture, the [Sal]− showed significantly higher degradation tendency in comparison with [bmim]+.
 | ||
| Fig. 6 Photodegradation of [bmim]Cl and [Sal]Na (c0 = 0.38 mmol L−1) using SS radiation and TiO2 P25 (1.67 g L−1) along the time. | ||
3.5 Degradation in different water types
It was very important to study the stability properties of ionic liquids toward AOPs in different types of water, since it is very likely that these substances will get into the different types of water that can be often found in the environment.The studied degradation processes included heterogeneous Fenton and photo-Fenton processes. Fig. 7 depicts the degradation curves of the ionic liquid in different waters by applying dark heterogeneous Fenton process. In the case of [bmim]+ (Fig. 7a), experimental data obtained after 180 min indicated that efficiency of degradation decreases in the following order: pond < rain < tap < river < condensate water, showing a strong dependence on the constitution of the radiated media.
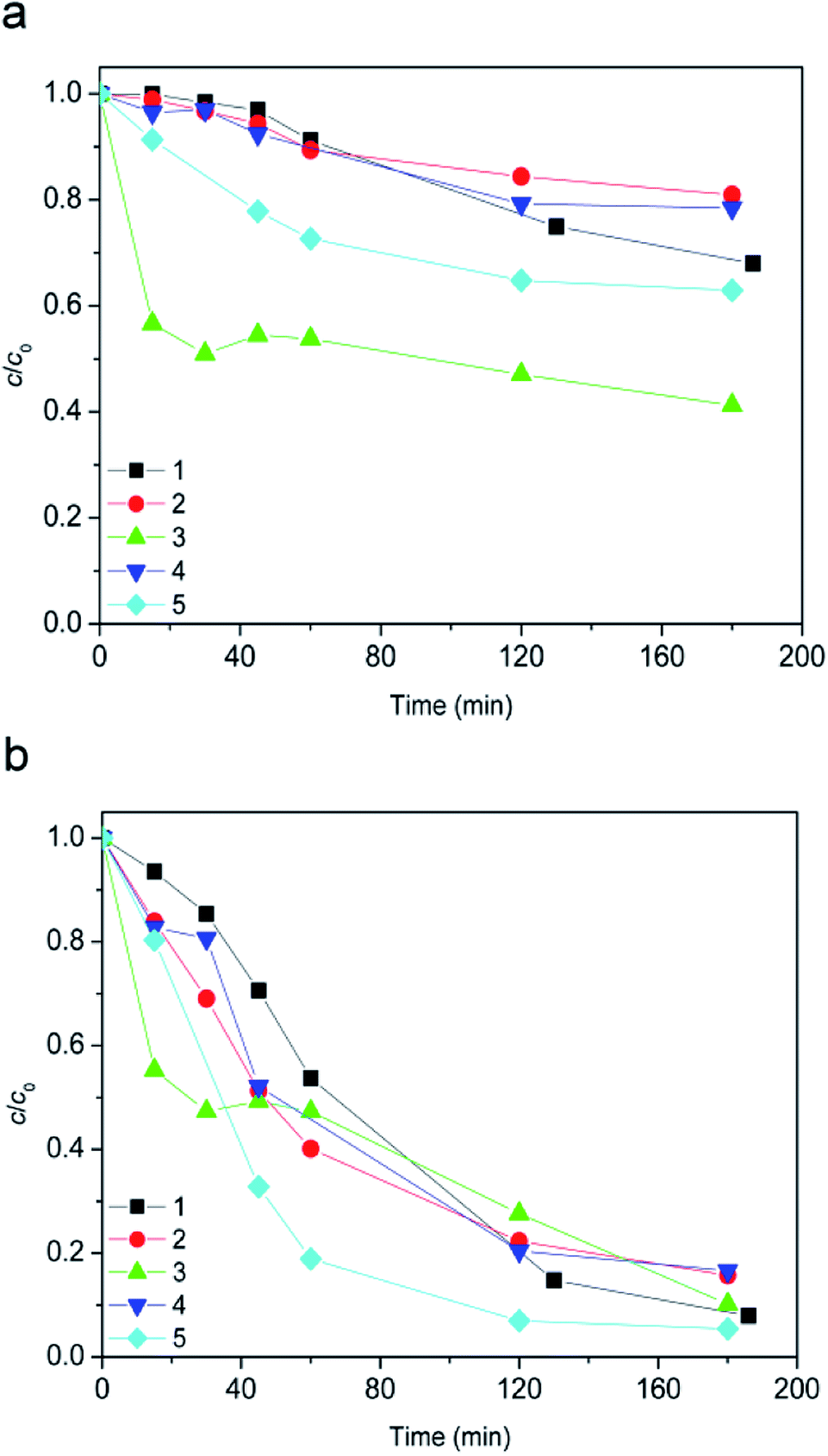 | ||
| Fig. 7 Evolution of ionic liquid (c0 = 0.38 mmol L−1) dark conversion along the time: (a) [bmim]+ and (b) [Sal]−. Water matrix: (1) tap water; (2) condensate water; (3) pond water; (4) river water; (5) rain water. Other reaction conditions: 7.2Fe/TiO2 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8. | ||
During the [bmim]+ degradation in acidic media (pH 2.8) the surface of 7.2Fe/TiO2 particles was positively charged, so that the extent of adsorption of metal ions was relatively insignificant. From the literature it is known that Na+, K+, Ca2+, and Mg2+ ions are the common cations in aquatic environment. They are all in the highest and stable oxidation state and cannot capture electrons or holes in solution.40 In the case of dark Fenton process it can be assumed that the [bmim]+ degradation efficiency mostly depended on the P, S, and Cl content (Table S1, ESI†). The highest degradation efficiency was found in pond water and it is probably due to the lowest total content of P, S, and Cl. By comparison of composite of pond water with rainwater in which the efficiency of the [bmim]+ degradation is some lower, it can be seen that in rainwater was 4 times higher content of P, 1.5 times higher S content, but 1.3 times lower Cl content. In the case of tap water also in comparison with pond water, the S content was 8 times lower, but the contents of P and Cl were eight and two times higher, respectively.
Furthermore, the Danube water had the highest content of P, S and Cl, so its degradation efficiency was the lowest. It can be assumed that the impact of content on the efficiency of degradation process decreases in the following order: P > Cl > S. Bali et al. came to a similar conclusion of negligible effect of SO42− and Cl−, in comparison with the effect of PO43− which was remarkable.41 Condensate water has the lowest P content, followed by pond water. The lower efficiency of degradation observed in condensate water can be attributed to the highest content of copper which leads to complexation with humic acids.42 On the other hand, in the case of [Sal]− after 180 min of degradation approximately equal efficacy was observed for all types of used water (Fig. 7b).
By applying a dark Fenton process, for all types of investigated water, a higher degradation efficiency of [Sal]− compared to [bmim]+ was observed (Fig. 7bvs.a). This can be explained by the fact that salicylic acid could present in different forms – non-dissociated at pH < 3.0, and dissociated at pH 6.0–12 (the pKa for salicylic acid is 2.97).43 It was also found that the adsorption of salicylate was the highest at pH 3 on iron containing surface.44 Besides, higher degradation efficiency as recorded in our case, can be explained by strong ability of salicylic acid to complex with surface iron atoms of photocatalyst under acidic conditions.32
Further studies were focused on examining of ionic liquids degradation efficiency in different types of water by using heterogeneous photo-Fenton process (Fig. 8). For both [bmim]+ and [Sal]− a significant increase in the degradation efficiency by SS/7.2Fe/TiO2/H2O2 process was observed (Fig. 5vs.8). The small contribution to degradation efficiency comes from the heterogeneous photo-Fenton process (see simulated sunlight photodegradation studies). However, significantly greater contribution to degradation efficiency comes from presence of SS radiation due of dissolved organic carbon (DOC). By absorption of sunlight radiation DOC could provide a rich variety of photochemical reactions.45 The resulting excited states of the DOC could participate in energy transfer, electron transfer, and free radical reaction, which affect the fate of aquatic pollutants.46
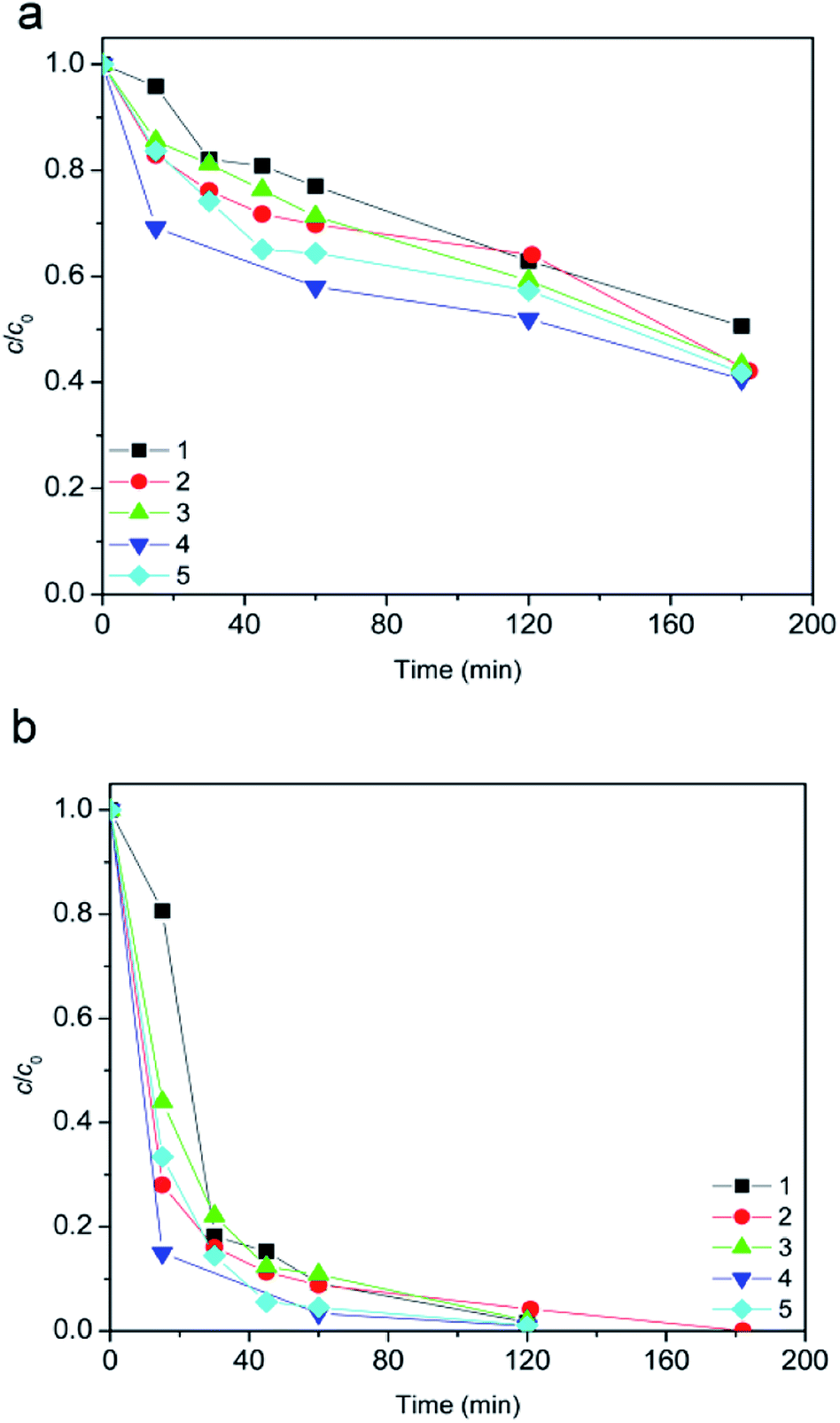 | ||
| Fig. 8 Evolution of ionic liquid (c0 = 0.38 mmol L−1) degradation along the time using SS radiation: (a) [bmim]+ and (b) [Sal]−. Water matrix: (1) tap water; (2) condensate water; (3) pond water; (4) river water; (5) rain water. Other reaction conditions: 7.2Fe/TiO2 (1.67 g L−1) + 45 mmol L−1 H2O2 + 70 μL h−1 H2O2 at pH 2.8. | ||
In this case, it can be concluded that the present DOC together with SS radiation provided a powerful sensitization effect (DOC was significant source of OH radicals production) and thus increase the ionic liquid degradation efficiency. At the end it can also be concluded that the amount range and composition of DOC (Table 1) in different types of water was such that the ionic liquid degradation efficiency in presence SS were nearly the same.
3.6 Identification of intermediates
Identification of the intermediates was carried out using the HPLC-ESI-MS/MS technique. Based on their MS/MS fragmentation data, twenty-two intermediates originating from [bmim]+ were identified in positive mode, and one intermediate originating from [Sal]− was identified in negative mode (Table 2; S2, ESI†).| Peak | t R (min) | M mi | Molecular formula | Structure |
|---|---|---|---|---|
| [bmim]+ | 2.88 | 139 | C8H15N2 |
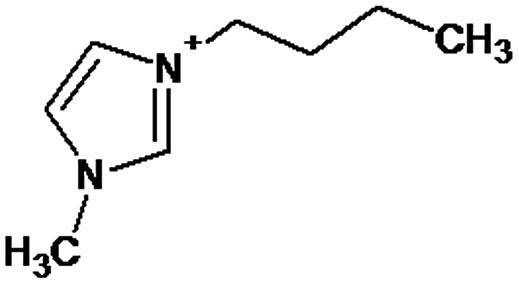
|
| P1 | 2.58 | 137 | C8H13N2 |
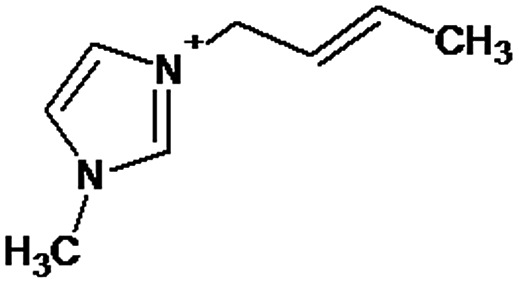
|
| P2 | 2.41 | 153 | C8H13N2O |
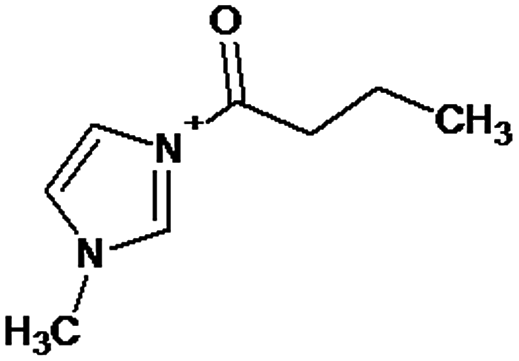
|
| P3 | 2.54 | 153 | C8H13N2O |

|
| P4 | 2.37 | 155 | C8H15N2O |

|
| P5 | 2.47 | 155 | C8H15N2O |

|
| P6 | 2.55 | 171 | C8H15N2O2 |
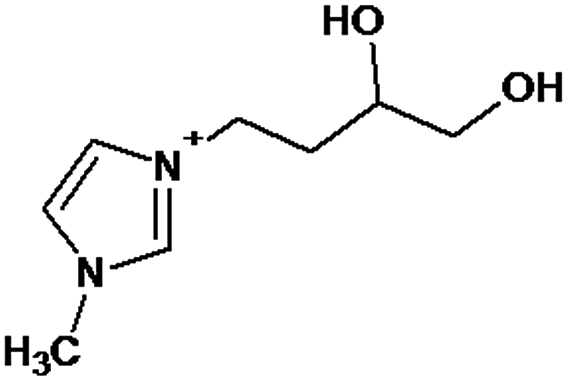
|
| P7 | 2.76 | 171 | C8H15N2O2 |

|
| P8 | 2.82 | 173 | C8H17N2O2 |
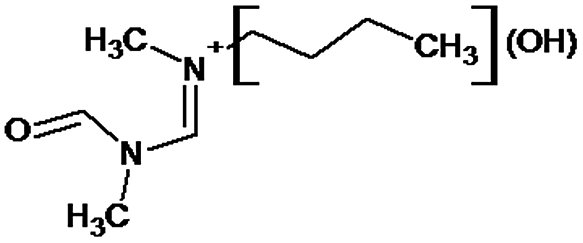
|
| P9 | 2.92 | 173 | C8H17N2O2 |
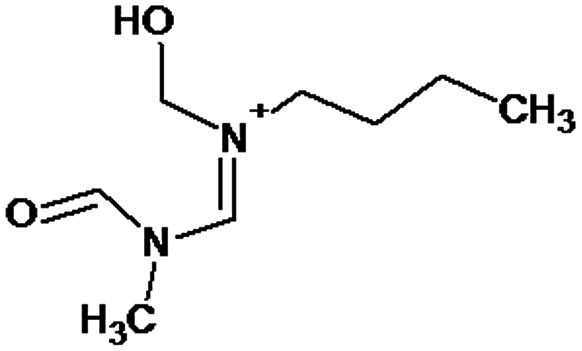
|
| P10 | 2.30 | 187 | C8H15N2O3 |
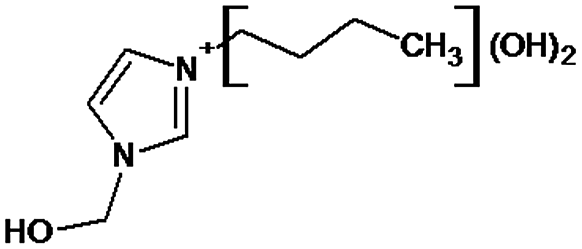
|
| P11 | 2.41 | 187 | C8H15N2O3 |
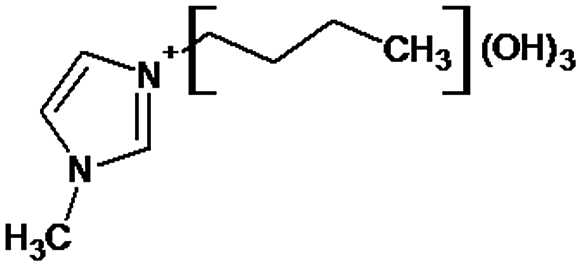
|
| P12 | 2.73 | 187 | C8H15N2O3 |
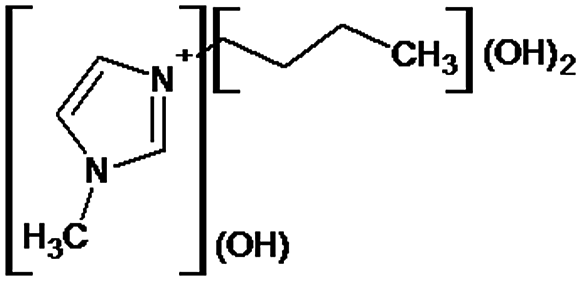
|
| P13 | 2.36 | 189 | C8H17N2O3 | — |
| P14 | 3.23 | 189 | C8H17N2O3 |
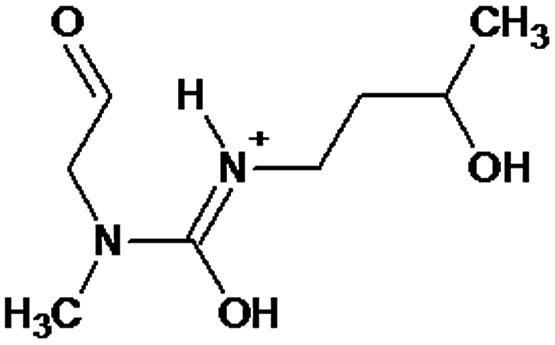
|
| P15 | 2.13 | 203 | C8H15N2O4 |

|
| P16 | 2.20 | 203 | C8H15N2O4 |

|
| P17 | 2.47 | 203 | C8H15N2O4 |

|
| P18 | 2.57 | 203 | C8H15N2O4 |

|
| P19 | 2.67 | 203 | C8H15N2O4 |

|
| P20 | 2.49 | 205 | C8H17N2O4 | — |
| P21 | 2.63 | 205 | C8H17N2O4 | — |
| P22 | 2.79 | 205 | C8H17N2O4 | — |
| [Sal]− | 9.60 | 137 | C7H5O3 |

|
| P23 | 7.75 | 153 | C7H5O4 |

|
P1 is a compound with Mmi = 137, which is 2 mass units lower than the molecular mass of 3-butyl-1-methylimidazolium (PI), indicating loss of two hydrogen atoms, most likely from butyl-chain, indicating that P1 is 3-(2-but-2-en-1-yl)-1-methyl-1H-imidazol-3-ium. P2 and P3 are ionic compounds with Mmi = 153. ΔMmi of 14 indicates the presence of oxo group in both compounds, thus it seems likely that P2 is 3-butanoyl-1-methyl-1H-imidazol-3-ium and P3 is 1-methyl-3-(2-oxobutyl)-1H-imidazol-3-ium. P4 and P5 are ionic compounds with Mmi = 155. Mass difference of 16 units suggests hydroxylation, with compound P4 likely standing for 3-(1-hydroxybutyl)-1-methyl-1H-imidazol-3-ium, and compound P5 being 3-(4-hydroxybutyl)-1-methyl-1H-imidazol-3-ium. P6 and P7 are isobaric compounds with Mmi = 171, which is 32 units higher than the monoisotopic mass of 3-butyl-1-methylimidazolium (PI), indicating the presence of two hydroxyl groups. P6 could possibly represent 3-(3,4-dihydroxybutyl)-1-methyl-1H-imidazol-3-ium and P7 is 3-(2,3-dihydroxybutyl)-1-methyl-1H-imidazol-3-ium. Compounds P8 and P9 both have Mmi = 173. The monoisotopic mass, 34 mass units greater than that of 3-butyl-1-methylimidazolium, points out to the presence of two hydroxyl groups and either reduction of one double bond in imidazolium ring or ring cleavage. P8 likely stands for [formyl(methyl)amino]-N-(hydroxybutyl)-N-methylmethaniminium and P9 is likely N-butyl[formyl(methyl)amino]-N-(hydroxymethyl)methaniminium. Although compounds P10, P11 and P12 have identical monoisotopic masses of Mmi = 187, their fragmentation patterns suggest that they have various distribution of three hydroxyl groups (Δm/z = 48) present in them. P10 likely stands for 3-(dihydroxybutyl)-1-(hydroxymethyl)-1H-imidazol-3-ium, intermediate P11 was identified as 1-methyl-3-(trihydroxybutyl)-1H-imidazol-3-ium and P12 is 3-(dihydroxybutyl)-1-(hydroxymethyl)-1H-imidazol-3-ium. P13 and P14 both correspond to compounds with monoisotopic mass Mmi = 189, 50 mass units higher than the molecular mass of 3-butyl-1-methylimidazolium. Based on their PI MS2 spectra and specific patterns of fragmentation, it was concluded that these compounds could have either three hydroxyl groups and reduction of one double bond in imidazolium ring, or cleavage of imidazolium ring with hydroxyl and oxo group attached, along with another hydroxyl on butyl or N-methyl moiety. It was not possible to elucidate the structure of P13, while P14 most likely stands for hydroxy-N-(3-hydroxybutyl)[methyl(2-oxoethyl)amino]methaniminium. P15, P16, P17, P18 and P19 all have monoisotopic masses of Mmi = 203, indicating that they either have four additional hydroxyl groups in the 3-butyl-1-methylimidazolium structure, or cleaved imidazolium ring with two hydroxyl and two oxo groups attached. P15 corresponds either to N-(1,3-dihydroxybutyl)-N-formyl[formyl(methyl)amino]methaniminium, or 3-(1,3-dihydroxybutyl)-4,5-dihydroxy-1-methyl-1H-imidazol-3-ium. P16 and P17 are both most likely tetrahydroxy derivatives of 3-butyl-1-methylimidazolium. Fragmentation patterns of P18 and P19 are very similar, with the only difference in the positions of the four hydroxyls, which cannot be determined with certainty. P20, P21 and P22 all correspond to compounds with monoisotopic mass Mmi = 205, which is 2 units higher than compounds P15–P19. This leads to an assumption that these compounds have four hydroxyls and one reduced double bond in imidazolium ring, or two hydroxyls in butyl moiety and hydroxyl and oxo group bound to a cleaved imidazolium ring. PI MS2 spectra of P20 and P21 are almost identical, thus it was not possible to further elucidate their structures. There is also limited information from the fragmentation pattern for identification of the intermediate P22. P23 is a compound with m/z = 153, identified in the NI mode. Its mass is 16 units higher than salicylate, indicating that one hydroxyl more is present in this molecule. However, it is not possible to determine whether P23 stands for 3-hydroxysalicylate or 5-hydroxysalicylate.
3.7 Photocatalytic degradation of various ionic liquids by the SS/7.2Fe/TiO2/H2O2 process
Bearing in mind that the photo-Fenton process involving solar radiation has significant potential applications, we investigated the efficiency of the 7.2Fe/TiO2/H2O2 process in the presence of SS radiation on five various types of ionic liquids. Based on the obtained results, it may be concluded that all the ionic liquids were decomposed under these experimental conditions (Fig. S1, ESI†). For all studied ionic liquids it can be noted that the stability of salicylate anion was lower compared to their corresponding cation. Also, it can be observed that salicylate photodegradability in relation to the cation increased in the following order: [bmim]+ < [TBA]+ < [im]+ < [Hpmim]+. From the same figure we can also conclude that [im]+ cation in a smaller extent accelerates the degradation of salicylate compared with acyclic tetrabutylammonium cation 95.2% vs. 92.3% degradated, respectively. On the other hand, the introduction of methyl and butyl groups at the positions N-3 and N-1 of imidazolium ion decreases the efficiency of degradation for 32.3%, compared to that with a [im]+ cation alone. One might also note that the salicylate completely decomposed in 60 min, if only a terminal methyl group of the side alkyl chain is replaced by a hydroxyl group. Compared with other studied cations, maximum efficiency degradation has been reached in the case of [Bzmim]+ cation (68.4%), even though this cation contains two aromatic ring. This points to the significant impact of the anion structure on the cation degradation efficiency, and vice versa. Similarly, (Section 3.4), the degradation efficiency of the [bmim]+ in the form of chloride was even 36% higher in comparison with [bmim]+ in the form of salicylate.4. Conclusions
After 93 days in studied pH range between pH 2.8 and 10.0, [bmim]+ was stable to hydrolysis. In contrast to cation, [Sal]− was less stable as the pH value increased. The following dark experiments showed that the ionic liquid was stable in the presence of hydrogen peroxide alone as well as in a combination of the TiO2 photocatalysts (TiO2 P25 or TiO2 support) and the peroxide. However, in the presence of hydrogen peroxide and 7.2Fe/TiO2 after 180 minutes 27% of [bmim]+ and 92% of [Sal]− has been degraded. This behavior can be attributed to the heterogeneous Fenton process, and within this process existence of surface bonded iron oxide was crucial. UVA photolysis experiments showed that no significant change in the [bmim]+ and [Sal]− concentration could be observed. Besides direct photolysis, the contribution of UVA radiation on the studied AOPs was decreased in the following order: UVA/TiO2 P25/H2O2 > UVA/TiO2 P25 > UVA/7.2Fe/TiO2/H2O2 > UVA/H2O2 > UVA. For the first time, the performance of different AOPs through degradation of ionic liquid under the simulated sunlight radiation was examined. Comparing obtained results, with those obtained for the same processes which were carried out in the dark, in all cases increasing efficiency can be observed. Also, for all of the studied processes, ionic liquid photodegradation efficiency was significantly lower in the presence SS radiation, compared to that in the presence of UVA radiation. By studying mutual facilitation of [bmim]+ and [Sal]− on the photodegradation efficiency, the impact of [Sal]− on the [bmim]+ degradation efficiency was much more pronounced.From a practical point of view, it was very important to study the stability properties of ionic liquids toward AOPs in different types of water. Applying dark heterogeneous Fenton process on [bmim]+ indicated that efficiency of degradation decreases in the following order: pond < rain < tap < river < condensate water showing a strong dependence on the constitution of the radiated media. On the other hand, in the case of [Sal]− approximately equal efficacy was observed for all types of used water. Also, for all types of water, a higher degradation efficiency of [Sal]− compared to [bmim]+ was observed. Furthermore, in presence of SS radiation the present DOC provided a powerful sensitization effect and thus increases the ionic liquid degradation efficiency.
LC-MS/MS analysis indicated that intermediates with one or more OH groups were produced during the photocatalytic degradation process. In addition, for the most efficient UVA/TiO2 P25/H2O2 system structure of nineteen intermediates are assumed.
Acknowledgements
This article was financially supported by the Ministry of Education, Science and Technological Development of Republic of Serbia under project contracts ON172042 and ON172012.References
- P. Stepnowski and A. Zaleska, J. Photochem. Photobiol., A, 2005, 170, 45 CrossRef CAS.
- D. Kralisch, A. Stark, S. Korsten, G. Kreisel and B. Ondruschka, Green Chem., 2005, 7, 301 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- J. D. Holbrey and K. R. Seddon, Clean Technol. Environ., 1999, 1, 223 CrossRef.
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567 CrossRef.
- D. J. Tempel, P. B. Henderson, J. R. Brzozowski, R. M. Pearlstein and H. Cheng, J. Am. Chem. Soc., 2008, 130, 400 CrossRef CAS PubMed.
- T. Predel and E. Schlücker, Chem. Eng. Technol., 2009, 32, 1183 CrossRef CAS.
- Q. X. Liu, S. Z. E. Abedin and F. Endres, Surf. Coat. Technol., 2006, 201, 1352 CrossRef CAS.
- M. Markiewicz, J. Hupka, M. Joskowska and C. Jugnickel, Physicochem. Probl. Miner. Process., 2009, 43, 73 CAS.
- R. Kawano, H. Matsui, C. Matsuyama, A. Sato, M. A. B. H. Susan, N. Tanabe and M. Watanabe, J. Photochem. Photobiol., A, 2004, 164, 87 CrossRef CAS.
- B. Weyershausen and K. Lehmann, Green Chem., 2005, 7, 15 RSC.
- W. L. Hough, M. Smiglak, H. Rodríguez, R. P. Swatloski, S. K. Spear, D. T. Daly, J. Pernak, J. E. Grisel, R. D. Carliss, M. D. Soutullo, J. H. Davis and R. D. Rogers, New J. Chem., 2007, 31, 1429 RSC.
- R. Ferraz, L. C. Branco, C. Prudêncio, J. P. Noronha and Ž. Petrovski, ChemMedChem, 2011, 6, 975 CrossRef CAS PubMed.
- P. C. A. G. Pinto, D. M. G. P. Ribeiro, A. M. O. Azevado, V. D. Justina, E. Cunha, K. Bica, M. Vasiloiu, S. Reis and M. L. M. F. S. Saraiva, New J. Chem., 2013, 37, 4095 RSC.
- L. T. Silveira, A. M. A. Liberatore, I. H. J. Koh, M. A. Bizeto and F. F. Camilo, J. Nanopart. Res., 2015, 17, 129 CrossRef.
- J. Jacquemin, J. Feder-Kubis, M. Zorębski, K. Grzybowska, M. Chorążewski, S. Hensel-Bielówska, E. Zorębski, M. Paluch and M. Dzida, Phys. Chem. Chem. Phys., 2014, 16(8), 3549 RSC.
- P.-Y. Chen and Y.-T. Chang, Electrochim. Acta, 2012, 75, 339 CrossRef CAS.
- V. M. Egorov, D. I. Djigalio, D. M. Mometenko, D. V. Chernyshov, I. I. Torocheshnikova, S. V. Smirnova and I. V. Pletnev, Talanta, 2010, 80, 1177 CrossRef CAS PubMed.
- L. Fischer, T. Falta, G. Koellensperger, A. Stojanovic, D. Kogelnig, M. Galanski, R. Krachler, B. K. Keppler and S. Hann, Water Res., 2011, 45, 4601 CrossRef CAS PubMed.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis and R. D. Rogers, Chem. Commun., 2001, 1(1), 135 RSC.
- A. P. de los Ríos, F. J. Hernández-Fernández, L. J. Lozano, S. Sánchez, J. I. Moreno and C. Godínez, J. Chem. Eng. Data, 2010, 55, 605 CrossRef.
- M. Czerwicka, S. Stolte, A. Müller, E. M. Siedlecka, M. Gołębiowski, J. Kumirska and P. Stepnowski, J. Hazard. Mater., 2009, 171, 478 CrossRef CAS PubMed.
- S. Stolte, S. Steudte, A. Igartua and P. Stepnowski, Curr. Org. Chem., 2011, 15, 1946–1973 CrossRef CAS.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351 CrossRef CAS.
- N. Banić, M. Vraneš, B. Abramović, J. Csanádi and S. Gadžurić, Dalton Trans., 2014, 43, 15515 RSC.
- B. F. Abramović, N. D. Banić and D. V. Šojić, Chemosphere, 2010, 81, 114 CrossRef PubMed.
- N. Banić, B. Abramović, J. Krstić, D. Šojić, D. Lončarević, Z. Cherkezova-Zheleva and V. Guzsvány, Appl. Catal., B, 2011, 107, 363 CrossRef.
- V. Guzsvány, N. Banić, Z. Papp, F. Gaál and B. Abramović, React. Kinet., Mech. Catal., 2010, 99, 225 Search PubMed.
- M. Pera-Titus, V. García-Molina, M. A. Baños, J. Giménez and S. Esplugas, Appl. Catal., B, 2004, 47, 219 CrossRef CAS.
- D. F. Laine and I. F. Cheng, Microchem. J., 2007, 85, 183 CrossRef CAS.
- S. Sabhi and J. Kiwi, Water Res., 2001, 35, 1994 CrossRef CAS PubMed.
- A. Goi, Y. Veressinina and M. Trapido, Chem. Eng. J., 2008, 143, 1 CrossRef CAS.
- M. Munoz, C. M. Domínguez, Z. M. de Pedro, A. Quintanilla, J. A. Casas and J. J. Rodriguez, Catal. Today, 2015, 240, 16 CrossRef CAS.
- E. M. Siedlecka, A. Więckowska and P. Stepnowski, J. Hazard. Mater., 2007, 147, 497 CrossRef CAS PubMed.
- A. Pieczyńska, A. Ofiarska, A. F. Borzyszkowska, A. Białk-Bielińska, P. Stepnowski, S. Stolte and E. M. Siedlecka, Sep. Purif. Technol., 2015, 156, 522 CrossRef.
- E. M. Siedlecka, S. Stolte, M. Gołębiowski, A. Nienstedt, P. Stepnowski and J. Thöming, Sep. Purif. Technol., 2012, 101, 26 CrossRef CAS.
- N. Tomić, M. Grujić-Brojčin, N. Finčur, B. Abramović, B. Simović, J. Krstić, B. Matović and M. Šćepanović, Mater. Chem. Phys., 2015, 163, 518 CrossRef.
- S. S. Lin and M. D. Gurol, Environ. Sci. Technol., 1998, 32, 1417 CrossRef CAS.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1 CrossRef CAS.
- C. Wang, L. Zhu, M. Wei, P. Chen and G. Shan, Water Res., 2012, 46, 845 CrossRef CAS PubMed.
- U. Bali, E. Ç. Çatalkaya and F. Şengül, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2003, 38, 2259 CrossRef.
- M. Kamiya and K. Kameyama, Chemosphere, 2001, 45, 231 CrossRef CAS PubMed.
- INCHEM IPCS, Salicylic Acid, http://www.inchem.org/documents/pims/pharm/pim642.htm, 2.2.2016.
- J. Krýsa, J. Jirkovský, O. Bajt and G. Mailhot, Catal. Today, 2011, 161, 221 CrossRef.
- M. Rejto, S. Saltzmann, A. J. Acher and L. Muszkat, J. Agric. Food Chem., 1983, 31, 138 CrossRef CAS.
- R. A. Larson and E. J. Weber, Reaction Mechanisms in Environmental Organic Chemistry, CRC Press, Boca Raton, FL, 1994 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Proposed structures of intermediates, ICP-MS scan signal intensities, and photodegradation of different ionic liquids. See DOI: 10.1039/c6ra04416f |
| This journal is © The Royal Society of Chemistry 2016 |