
Tunable emission color and mixed valence state via the modified activator site in the AlN-doped Sr3SiO5:Eu phosphor†
Abstract
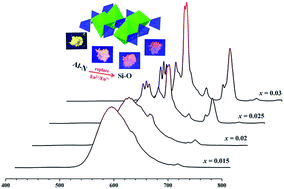
Commercially white LEDs have cool white light and a low color rendering index (CRI) due to the absence of red light emission. Considering the respective characteristic luminescence properties of Eu2+ and Eu3+ ions, the combination of the two activators into a single host compound shows promisingly technical feasibility to realize warm w-LEDs with a high CRI but it is still a challenge in practice. Here, we confirmed the mixed europium valence states in Sr3Si1−xAlxO5−2xNx:Eu phosphors, synthesized by a solid-state reaction method under a H2/N2 atmosphere. The crystal structure was characterized by X-ray diffraction and found to crystallize in the Sr3SiO5 phase. The samples show broad excitation bands (250–400 nm) and tunable emission colors from yellow to red, and the CIE chromaticity coordinates of Sr2.97Si1−xAlxO5−2xNx:0.03Eu (x = 0, 0.03, 0.2) were (0.518, 0.474), (0.607, 0.385) and (0.630, 0.367), respectively. The modification of activator sites caused by Al3+–N3− replacing Si4+–O2− explained the coexistence of Eu2+/Eu3+ ions, which was demonstrated by Rietveld refinement, photoluminescence analysis and fluorescence lifetime studies. Furthermore, the obtained phosphors also possessed good thermal stabilities. These results suggest that the Sr3Si1−xAlxO5−2xNx:Eu phosphors show potential applications in w-LEDs.
 Please wait while we load your content...
Please wait while we load your content...

