
LPA1 extracellular loop residues 115 and 191 are not required for receptor activation but prevent Ki16425 super-antagonism†
Abstract
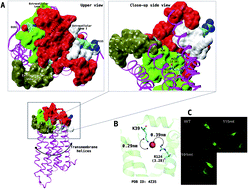
Lysophosphatidic acid receptor 1 (LPA1) is a clinically important target, which confers a biological response to lysophosphatidic acid-type agonists. Recently, the 3D structure of LPA1 resolved using X-ray crystallography and our site-directed mutagenesis has provided significant insight into the mechanism of LPA1 activation and antagonism. In the present study, we report that extracellular loop residues (R115 and D191) are not required for receptor activation but repress Ki16425-type super-antagonism but not LPA-analogue antagonists using a combination of site-directed mutagenesis and intracellular calcium assay procedures. The underlying mechanism was further queried using an all-atom molecular dynamics (MD) simulation. The result showed that Ki16425 when bound to LPA1 evolved characteristic conformational and optimal route communication signatures as a LPA (C18:1)-agonist which is reversed in R115A and D191A mutants. In conclusion, Ki16425 is a super-antagonist when bound to mutant (R115A and D191A) LPA1.
 Please wait while we load your content...
Please wait while we load your content...

