
Chemical components from the seeds of Catalpa bungei and their inhibitions of soluble epoxide hydrolase, cholinesterase and nuclear factor kappa B activities†
Abstract
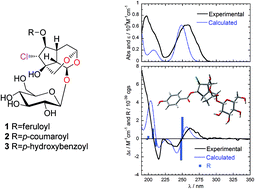
Two new chlorinated iridoids, named bungosides A (1) and B (2), were isolated from the seeds of Catalpa bungei (family Bignoniaceae). Their structures were elucidated on the basis of NMR analysis. Twenty known compounds (3–22) were also characterized, including in one case (6-O-p-hydroxybenzoylglutinoside, 3) the assignment of the absolute configuration by employing electronic circular dichroism (CD) and time-dependent density functional theory (TDDFT) calculations. Compounds 1–3 were unusual cage-like iridoids with an oxygen bridge between C-3 and C-10. Among the isolates, ursolic acid lactone (14) was the most active soluble epoxide hydrolase (sEH) inhibitor with an IC50 19.1 ± 0.8 μM. In addition, D-pinoresinol (19) and ladanein (21) displayed selectively inhibitory effects on butyrylcholinesterase (BChE) with IC50 of 31.5 ± 0.4 μM and 33.0 ± 2.3 μM, respectively, but not acetylcholinesterase (AChE) activity, and only compound 18 suppressed NF-κB activity in HepG2 cells (IC50 21.53 ± 2.37 μM).
 Please wait while we load your content...
Please wait while we load your content...

