
Understanding the structural requirements of cyclic sulfone hydroxyethylamines as hBACE1 inhibitors against Aβ plaques in Alzheimer's disease: a predictive QSAR approach†
Abstract
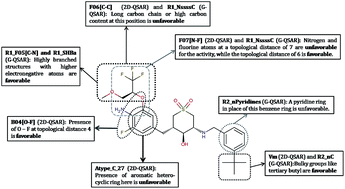
Beta (β)-site amyloid precursor protein cleaving enzyme 1 (BACE1) is one of the most important targets in Alzheimer's disease (AD), which is responsible for production and accumulation of beta amyloid (Aβ). The Aβ plaques are one of the major hallmarks, which are believed to be the fundamental cause of AD. Thus, a rational treatment strategy is to find efficient BACE1 inhibitors against AD. In the present study, we have developed two quantitative structure–activity relationship (QSAR) models (viz. two dimensional-QSAR and group based-QSAR), employing 91 cyclic sulfone (or sulfoxide) hydroxyethylamines as human BACE1 (hBACE1) inhibitors to identify the important structural features that are responsible for their activity. To our knowledge, this is the first QSAR report for this chemical category of BACE1 inhibitors. The developed models were stringently validated utilizing various validation metrics including the recently proposed mean absolute error (MAE)-based criteria. Based on this study, we observed that MAE-based criteria are the most appropriate way to determine the true prediction quality of the models. Unlike R2-based metrics, the MAE-based criteria were found to provide an unbiased judgment irrespective of the response range and/or distribution of response values around the training/test mean. The mechanistic interpretation of each model was performed in detail, while focusing on the important substructures that were found favorable or unfavorable for hBACE1 activity.
- This article is part of the themed collection: Towards understanding and treating Alzheimer’s disease
 Please wait while we load your content...
Please wait while we load your content...

