
Wheat germ agglutinin modification of lipid–polymer hybrid nanoparticles: enhanced cellular uptake and bioadhesion†
Ying Liu‡
,
Yiqing Zhao‡,
Jinguang Liu,
Meiying Zhang,
Minglei Yu and
Nianping Feng*
Department of Pharmaceutical Sciences, School of Pharmacy, Shanghai University of Traditional Chinese Medicine, 1200 Cailun Road, Zhangjiang Hi-Tech Park, Pudong New District, Shanghai 201203, PR China. E-mail: npfeng@hotmail.com; npfeng@shutcm.edu.cn; Fax: +86 21 5132 2198; Tel: +86 21 5132 2198
First published on 5th April 2016
Abstract
Lipid–polymer hybrid nanoparticles (LPNs) have emerged as promising nanocarriers for oral delivery of poorly water-soluble drugs because they combine the advantages of polymeric and lipid-based nanoparticles. However, rational surface engineering of LPNs to reduce clearance in the gastrointestinal tract and improve bioavailability remains a challenge. Modification of LPNs with wheat germ agglutinin (WGA), a specific bioadhesive material, was hypothesised to increase site-specific adhesion and improve bioavailability. This study focused on the preparation and characterisation of WGA-modified LPNs (WGA-LPNs) for oral delivery of oridonin. WGA-LPNs were produced by incubating synthetic WGA-1,2-dioleoyl-sn-glycero-3-phosphoethanolamine with LPNs, which had been formed using nanoprecipitation. The WGA-LPNs had a particle size of 326.7 ± 5.2 nm and a zeta potential of −31.8 ± 1.04 mV. The core–shell structure and the WGA binding were confirmed by immune gold labelling electron microscopy. Approximately 80% of the drug was released from the nanoparticles within 24 h. WGA-LPNs showed efficient binding to both Caco-2 and HT29-MTX cells and underwent receptor-mediated endocytosis, as evidenced by cellular uptake and confocal imaging. In addition, coumarin 6-loaded WGA-LPNs showed enhanced uptake in the ligated intestinal loop model in vivo, probably due to improved bioadhesion to the villi. These results suggested that WGA-LNPs showed increased intestinal bioadhesion and cellular uptake and have the potential to improve the oral delivery of poorly water-soluble drugs.
1. Introduction
Oridonin (Fig. 1) is a naturally occurring diterpenoid active ingredient extracted from Rabdosia rubescens (Hamsl.) Hara. It has been shown to induce cancer cell death1–3 and exhibits remarkable anti-proliferative and anti-tumour effects, especially in esophageal, colorectal, pancreatic, and hepatic cancers.4,5 The molecular mechanisms involved in these effects include cell cycle regulation,4,6 induction of apoptosis and induction of autophagy.1,7 In addition, this compound has effective anti-bacterial and anti-inflammatory activities.8,9 Despite these beneficial properties, the extensive application of oridonin has been limited by its poor solubility, instability, low bioavailability, and rapid plasma clearance. In practice, even when patients receive large doses of crude extract tablets with low oridoin in content (6–15 tablets per day), bioavailability is variable owing to poor water solubility. Because of the need to improve the therapeutic effects, quality of life, and/or ease of follow-up therapy, substantial efforts have been made recently to overcome these physiochemical limitations and improve the in vivo behaviour of oridonin. These have included the use of self-microemulsifying drug delivery systems,10 nanostructured lipid carriers,11 and polymeric nanoparticles.12 These nanocarriers improved bioavailability by enhancing the physiochemical properties of oridonin, including its solubility, stability and/or permeability. Increasing the residence time in the gastrointestinal tract is an alternative strategy by oral route which would also be predicted to improve bioavailability. | ||
| Fig. 1 Chemical structure of oridonin. | ||
Lipid–polymer hybrid nanoparticles (LPNs) have attracted increasing interest from researchers investigating approaches to the delivery of poorly water-soluble drugs because of their unique combination of the advantages of liposomes and polymeric nanoparticles.13 Poorly water-soluble drugs can be loaded in the hydrophobic polymeric core and drug release is regulated by the lipid shell, enhancing nanoparticle stability and increasing their half-life in the systemic circulation.14–16 These features have been characterised in previous studies using oral, topical, parenteral or other routes of administration. LPNs have been used for oral delivery of low molecular weight drugs, such as vincristine sulphate.17 This achieved some improvement in oral bioavailability due to a high encapsulation rate, controlled drug release, and efficient cellular uptake. Although these studies indicated that LPNs had the potential to improve the oral bioavailability of poorly water-soluble drugs, few such systems have been reported and their development remains challenging.
Lectins are non-immunogenic proteins or glycoproteins that can bind to glycosylated membrane components. Wheat germ agglutinin (WGA) is a non-toxic plant lectin that has the capacity to bind N-acetyl-D-glucosamine and sialic acid on intestinal enterocytes and microfold cells. It has a stable structure that is resistant to enzymatic degradation and pH variation, especially under acidic conditions. Therefore, it has been used in oral micro/nanoparticle formulations to facilitate specific bioadhesion.18–20
One strategy employed to achieve lectin modification of nanoparticles is to include WGA in the formulation of conjugates, which are then used as components to prepare the nanoparticles.21 Another two chemical methods have been reported for WGA modification of macro/nanoparticles: glutaraldehyde and carbodiimide coupling.22,23 These chemical synthesis methods require specific functional groups, such as amino, hydroxyl, or carboxy groups. Although these methods achieved WGA modification of nanoparticles, further investigation is required to develop milder linkage reactions and to limit WGA deactivation. The post-insertion method is a physical strategy that incorporates ligands or conjugates into pre-formed carriers using a simple incubation step. It has been used to link ligands to many nanocarriers, including liposomes, lipid nanocapsules and lipid nanoparticles.24 The advantages of this method lie in the ease of operation, reduced deactivation and a controllable modification rate.25 This method has been used to decorate liposomes with a range of ligands, including proteins26 and polymeric conjugates.27 Post-insertion can be achieved by preparing ligand–polyethylene glycol (PEG)–lipid conjugates, and incubating these with preformed liposomes or lipid nanocapsules. Another simple post-insertion was efficiently conducted by inserting protein-1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) conjugates into the lipid bilayers of lipid nanoparticles.28 The present study, therefore, opted to use the latter to develop the preparations.
WGA-modified LPNs (WGA-LPNs) were prepared using post-insertion of WGA–DOPE into the lipid bilayers of preformed LPNs. Then, WGA-LPNs were characterised in vitro, and their bioactivities and release were evaluated. Previously, the cellular uptake behaviours of WGA-modified LPNs were illustrated,29 but the uptake mechanism was not completely investigated. Therefore, we examined the interactions between WGA-LPNs and cells and performed uptake studies in vitro using both Caco-2 and HT29-MTX cells. The goblet cell-like HT29-MTX cell line was adapted from the human colon cancer HT29 cell line and has mucus-secreting capacity.30 These cells have been used to mimic goblet cells in several studies investigating cellular nanoparticle uptake. Ex vivo bioadhesion studies have been performed that introduce the preparations into the isolated intestinal segment and then quantitatively determine the fluorescence intensity in intestinal segments.29 To simulate more closely the conditions of the intact animal intestine, in vivo absorption and bioadhesion was evaluated using a ligated intestinal loop model, and nanoparticle distribution was investigated in the villi.
2. Materials and methods
2.1. Materials
Oridonin (purity > 98%) was provided by Herbfine Co. (Jiangxi, China). Poly(lactic-co-glycolic acid) (PLGA; 50/50, intrinsic viscosity 0.47 dl g−1) was from Jinan Daigang Biomaterial Co. Ltd. (Shandong, China). WGA from Triticum vulgaris (wheat), 3-maleimidobenzoic acid N-hydroxysuccinimide ester crystalline (MBS), DOPE, 5,5′-dithiobis(2-nitrobenzoic acid) (Ellmann's Reagent), coumarin 6, 2-iminothiolane hydrochloride (Traut's Reagent), goat anti-WGA antibody, 10 nm colloidal gold-labelled rabbit anti-goat antibody, and Hoechst 33258 were obtained from Sigma-Aldrich Co. (MO, USA). Hoechst 33342 was obtained from Aladdin (Shanghai, China). Soya phosphatidylcholine (LIPOID S 100) was purchased from Libao Biological Technology Co. (Shanghai, China). 1,2-Distearoyl-sn-glycero-3-phosphoethanol-amine-N-methoxy(polyethyleneglycol)-2000 (DSPE-PEG2000) was purchased from AVT Technology Co. (Shanghai, China). Sepharose™ CL-4B was obtained from GE Healthcare Bio-Sciences Co. (Uppsala, Sweden). Dulbecco's modified Eagle's medium (DMEM, high glucose), foetal bovine serum (FBS, Australian origin), Hank's Balanced Salt Solution (HBSS), penicillin and streptomycin and 1 × trypsin–ethylene diamine tetraacetic acid (EDTA) solution (0.25% trypsin with 0.53 mM EDTA) were obtained from Gibco Life Technologies (Grand Island, NY, USA). NP40 cell lysis buffer was from Thermo Fisher Scientific (Invitrogen Novex, Carlsbad, CA, USA). Purified deionised water (Millipore, Bedford, MA) was used in all experiments. All other chemicals were of high-performance liquid chromatography (HPLC) or analytical grade.Caco-2 cells were kindly provided by Professor Hai Wei (Center of Chinese Medicine Therapy and Systems Biology, Shanghai University of TCM, Shanghai, China). HT29 cells were obtained from the China Center for Type Culture Collection (Wuhan, China). Caco-2 cells were cultured in DMEM containing 10% FBS, penicillin (100 IU ml−1), streptomycin (100 μg ml−1) and 1% non-essential amino acids at 37 °C in the presence of 5% CO2. HT29-MTX cells were adapted from HT29 cells and characterised as reported previously.30,31
Male Wistar rats weighing 200 ± 20 g were supplied by the Shanghai Laboratory Animal Center. The animal experiment protocols were approved by the Institutional Animal Care and Use Committee, Shanghai University of TCM. The rats were maintained under standard conditions for 1 week prior to the experiment.
2.2. Preparation and characterisation of LPNs
The LPNs were prepared using a modified nanoprecipitation technique.13 In brief, PLGA and oridonin were dissolved in acetonitrile (as the oil phase). The aqueous phase, containing the lipids pre-dissolved in ethanol (100 mg ml−1), was heated in a water bath at 65 °C to form a homogeneous dispersion. The oil phase was then poured into the aqueous phase under stirring at 100 rpm, followed by vortexing for 3 min. After stirring at 100 rpm for 2 h at room temperature, the acetonitrile was removed in a rotary evaporator. The resulting LPNs were filtered through a 0.8 μm nylon membrane filter, introduced into a dialysis bag and submerged in phosphate-buffered saline (PBS; pH 7.4) for 12 h to remove unincorporated drug. Coumarin 6-loaded LPNs were prepared using the same procedure, except that oridonin was replaced by coumarin 6. The LPNs particle size and zeta potential were determined using a Zetasizer nano-ZS 90 (Malvern, UK). Membrane fluidity was determined by fluorescence anisotropy analysis using a fluorescence spectrophotometer (Cary Eclipse, CA, USA) with Scan Software Version of 1.1(132), using coumarin 6 as the fluorescent probe.32 The encapsulation efficiency of oridonin in LPNs was determined using an Amicon Ultra-4 centrifuge filter (10 kDa molecular weight cut-off, Millipore, Billerica, MA, USA), as previously described.292.3. Synthesis of WGA–DOPE
Maleimidobenzoyl–DOPE (MB–DOPE) was prepared as described previously, with some modification.28 WGA–DOPE was synthesised using MB–DOPE and thiolated WGA (WGA-SH; Fig. 2). MBS (15 μM in trichloromethane) was reacted with 10 μM DOPE in the presence of 50 μl triethylamine in a total volume of 3 ml. The reaction mixture was continuously stirred at room temperature overnight prior to evaporating trichloromethane under nitrogen. Then, 6 ml of ethanol was added to dissolve the residue. The resulting solution was injected into the appropriate amount of deionised water to produce MB–DOPE dispersions. WGA-SH was produced by reacting the amine groups of WGA with Traut's reagent in a ring opening reaction. WGA was first dissolved in HEPES (1 mg ml−1, pH 8) before adding 10 volumes of Traut's reagent, followed by gentle agitation for 1 h at room temperature. The resulting solution was added to a desalting column and centrifuged at 1000g for 2 min to remove the free Traut's reagent. The filtrate contained WGA-SH. Finally, WGA–DOPE was produced by incubating MB–DOPE dispersions with WGA-SH at a molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
 | ||
| Fig. 2 Schematic diagram depicting the preparation of WGA-LPNs. | ||
2.4. Preparation of WGA-LPNs
WGA-LPNs were prepared by post-insertion. WGA–DOPE was incubated with the LPNs for 18 h at room temperature. Free WGA–DOPE was then removed using a Sepharose CL-4B column, eluted by PBS (pH 7.4). The WGA-LPNs fraction was collected for further use.2.5. Characterisation of WGA-LPNs
Droplet size, zeta potential and morphology were analysed as described above. The binding efficiency of WGA to LPNs was determined using a previously reported method.20Transmission electron microscopy (TEM) using immune gold labeling: WGA bound to the nanoparticle surface was observed by TEM after immune gold labeling.33 WGA-LPNs (1 mg) were incubated with 10 μl of rabbit anti-WGA primary antibody (1 mg ml−1) at 37 °C for 1 h, followed by removal of the free antibody using a Sepharose CL-4B column. The WGA-LPNs were then incubated with 50 μl of 10 nm colloidal gold-labelled goat anti-rabbit secondary antibody at 37 °C for 3 h. The free secondary antibody was removed using a Sepharose CL-4B column. The WGA-LPNs were collected, dropped onto a copper grid, stained with 1% phosphotungstic acid, and air-dried prior to observation by TEM (JEM 1230; JEOL, Tokyo, Japan).
2.6. In vitro oridonin release and distribution within LPNs
In vitro oridonin release from LPNs and WGA-LPNs was analysed using a dialysis bag diffusion method, with some modification. LPNs or WGA-LPNs were introduced into a pre-treated dialysis bag with a molecular weight cut-off of 14 kDa and immersed into the PBS (pH 7.4) release medium containing 1% Tween 80. The release medium was in a shaking water bath at a speed of 100 strokes per min at 37 ± 1 °C. At pre-determined time intervals (1, 2, 4, 8, 12, 24, and 48 h), the dialysis bag was taken out. The liquid in the dialysis bag was withdrawn and thoroughly mixed with the appropriate amount of ethanol and acetonitrile (1:1, v/v). Then, the samples were filtered through 0.45 μm membrane filters. The oridonin level was determined using HPLC (Agilent 1260; Agilent, Santa Clara, CA, USA) using a Platisil ODS column (C18; 250 mm × 4.6 mm, 5 μm; Dikma Technology, Shanghai, China). A mobile phase of methanol/water (60:40, v/v) was pumped at a flow rate of 1.0 ml min−1. The detection wavelength was 238 nm.
We determined the distribution of oridonin between the lipid shell and polymeric core of the LPNs. Oridonin-loaded LPNs were placed in a dialysis bag, which was then covered by PEG20000 to remove aqueous liquids and free drug. One-hundred microlitres of the concentrated LPNs were divided into two equal parts. One part was mixed with 1 ml demulsifier (acetonitrile:ethanol (1:1, v/v)) and the other was mixed with 1 ml absolute ethanol. The concentration of oridonin in the demulsifier represented the level of oridonin in LPNs and the concentration in absolute ethanol represented the level in the lipid layer.
2.7. Cell uptake
Cellular uptake was examined using Caco-2 enterocyte cells and the goblet cell-like HT29-MTX cells. These cells were seeded into 96 well plates at a density of 1.5 × 104 cells per well and cultured for 7 days. The cells were rinsed three times with HBSS and then incubated in HBSS for 30 min at 37 °C. HBSS was removed and the cells were incubated with 100 μl fresh DMEM containing coumarin 6 loaded LPNs and WGA-LPNs (0.8 μg ml−1 of coumarin 6) at 37 °C for the indicated time-period. The media was then removed and the cells were rinsed twice with ice-cold PBS prior to the addition of 100 μl cell lysis buffer. The lysates were transferred to a tube and centrifuged at 12000 rpm (4 °C) for 10 min. The supernatant was collected and used to determine the fluorescence and the protein content. The fluorescence of coumarin 6 was determined using a fluorescence spectrometer (Synergy 2 Reader; Biotek, USA) with an excitation wavelength of 467 nm and an emission wavelength of 505 nm. The protein level was determined using a bicinchoninic acid assay kit (Thermo Fisher Scientific, Rockford, USA).
To investigate the potential cellular uptake pathways, the cells were pre-treated with three different inhibitors at 37 °C for 30 min.34 These inhibitors were 100 mM sodium azide (to deplete cellular ATP),35 10 μg ml−1 chlorpromazine (to inhibit the formation of clathrin vesicles) and 5 mM N-acetyl-D-glucosamine (to inhibit WGA binding to Caco-2 cells). After this pre-treatment, the cells were further incubated with coumarin 6 loaded LPNs and WGA-LPNs (0.8 μg ml−1 of coumarin 6) for 3 h. The media was then removed and the cells were washed twice with PBS and treated as described above prior to determinations of the lysate fluorescence and protein concentration. Cells exposed to LPNs or WAG-LPNs in the absence of inhibitors were used as controls. The cellular uptake by the control cells was defined as 100% and the uptake in the other groups was expressed relative to the control group.
2.8. Confocal laser scanning microscopy
Caco-2 and HT29-MTX cells (9:1) were cultured on glass-bottomed dishes until they reached approximately 50% confluence. The cells were then rinsed with PBS (pH 7.4) and incubated in 1.5 ml fresh culture medium containing 100 μl dispersions of LPNs or WGA-LPNs (0.8 μg ml−1 of coumarin 6) at 37 °C for 0.5, 1, or 2 h. The cells were then rinsed with PBS (pH 7.4) twice, fixed with 4% paraformaldehyde at room temperature for 20 min and then rinsed twice with PBS. The cells were stained with 10 μg ml−1 Hoechst 33258 fluorescent dye for 20 min and washed twice using PBS before observation under a confocal microscope (TCS SP2; Leica, Mannheim, Germany).
2.9. Bioadhesion studies in ligated intestinal loops
The ligated intestinal loop model was used to evaluate the absorption and bioadhesion of LPNs and WGA-LPNs in the rat jejunum.36 Rats were fasted overnight with free access to water. They were anaesthetised using 20% urethane, administered by intraperitoneal injection, and remained anaesthetised throughout the procedure. An approximately 2-inch incision was made on the midline of the abdomen. A 3 cm section of the jejunum was excised and the residual material, including excess food particles, was removed using pre-warmed Krebs–Ringer's solution at 37 °C. One end of the jejunal section was then ligated. The preparations (coumarin 6-loaded LPNs or coumarin 6-loaded WGA-LPNs, 0.8 μg ml−1 of coumarin 6) were injected carefully via the other end of the section, which was then ligated firmly. The intestinal loop was placed in the body cavity of the animal. At pre-determined times (1, 2 or 3 h), the rats were sacrificed and the loop was removed and washed with Krebs–Ringer's solution. After fixation by 4% paraformaldehyde for 3 h, the samples were embedded in Tissue-Tek optimum cutting temperature compound (Sakura Finetek USA Inc., Torrance, CA, USA) and snap-frozen in liquid nitrogen prior to cutting into thin sections on an ultra microtome (MicromHM525; Microm International, Walldorf, Germany). The sections were placed on microscope slides and rinsed three times with PBS. They were stained with 2% Hoechst 33342 for 15 min, followed by rinsing twice. The excess liquid was removed using filter paper and the sample was mounted using fluorescence mounting medium prior to observation using a Leica Microsystems TCS SP8 confocal microscope.2.10. Statistical analysis
Data were expressed as the mean ± standard deviation and analysed using one-way analysis of variance (ANOVA) with Tukey's post-test. Statistical significance was set as a p value of <0.05.3. Results and discussion
3.1. Preparation and characterisation of LPNs
In this study, a modified one-step nanoprecipitation method was used to form oridonin-loaded LPNs because this process is simpler and faster than the emulsification–solvent–evaporation method, making it more suitable for large-scale production.13,37 Particle size and surface charge are important biopharmaceutical features of nanoparticles. Formulation factors such as the lipid:polymer ratio and the organic phase:aqueous phase volume ratio were previously reported to influence the particle size and surface charge of LPNs.38 We therefore investigated the influence of the lipid:PLGA ratio on the particle size and encapsulation efficiency. As shown in Fig. 3A, LPNs generated using each lipid/polymer ratio showed desirable particle sizes, which ranged from 138–202 nm, with zeta potentials between −27.5 mV and −31.25 mV. The encapsulation rate increased in proportion to the lipid ratio. The majority of previous LPN studies focused on the influence of the lipid:PLGA ratio on particle size and encapsulation efficiency, as described above. In addition, the present study determined the membrane fluidity of the LPNs by fluorescence anisotropy analysis using coumarin 6. This was analysed to investigate whether lipid variations altered the physiochemical features of the lipid shell. As shown in Fig. 3B, there was a marked difference between the anisotropy values for LPNs prepared at ratios of 1:2 and 1:3, indicating decreased membrane fluidity as the lipid ratio increased. A lower membrane fluidity promotes drug encapsulation and hinders drug release. These findings indicated that the optimal lipid/PLGA ratio was 1:2. Our analysis of the organic phase/aqueous phase ratio indicated that higher ratios resulted in smaller LPNs. This was probably due to the requirement for sufficient contact between these two phases to facilitate LPN formation, in addition to modulating drug loading and encapsulation efficiency.39 Based on these findings, the organic:aqueous phase ratio was set at 1:5. Finally, we studied the influence of the initial drug input on particle size, zeta potential, drug loading and encapsulation rate. As shown in Fig. 3C, as the initial drug input increased, drug loading increased, while the encapsulation rate decreased significantly.
 | ||
| Fig. 3 Influence of formulation factors on particle size, zeta potential, encapsulation efficiency, drug loading and membrane fluidity of the lipid polymeric nanoparticles (LPNs). | ||
Based on these results, the final formulation composition (w/w) was as follows: 3.2% oridonin, 64.5% PLGA and 32.3% lipids. The resulting LPNs had a mean particle size of 203.9 nm, a zeta potential of −29.4 mV and an encapsulation efficiency of 81.83%. TEM images (Fig. 4A) showed that these were almost spherical, with a clear shell–core structure.
 | ||
| Fig. 4 Characterisation of nanoparticles, showing immune gold electron microscopy of (A) LPNs and (B) WGA-LPNs (×80000). (C) Haemagglutination test of WGA-LPNs in a red blood cell suspension, observed by optical microscopy. | ||
3.2. Preparation and characterisation of WGA-LPNs
The present study used post-insertion to generate WGA-LPNs that were 326.7 ± 5.2 nm in size, with a zeta potential of −31.8 ± 1.04 mV. The binding efficiency of WGA to LPNs was determined to be 27.5%. The binding of WGA on the surface of LPNs was verified using immune gold TEM with a primary anti-WGA antibody and a 10 nm gold-labelled secondary antibody. As shown in Fig. 4B, gold was observed near the WGA-LPNs surface and in the lipid layer, indicating the presence of WGA on these nanoparticles.To test the bioactivity of WGA in the preparations, haemagglutination tests were performed. As shown in Fig. 4C, WGA-LPNs induced significant erythrocyte aggregation, while incubation of erythrocytes with LPNs produced similar findings as incubation with PBS (ESI Fig. S1†). These results indicated that WGA retained its bioactivity following conjugation to the LPNs.
DSC analysis was performed to characterise the LPNs, to investigate oridonin entrapment and to study their interaction with WGA. Oridonin showed a characteristic peak at 256.9 °C (Fig. 5), and although this peak was retained in the physical mixture, its position was changed. This phenomenon has been demonstrated in other DSC studies examining the physical mixture of a drug with lipids.40,41 This change was considered to be due to the interaction between the drug and the lipid materials upon mixing, which decreased crystallinity and lowered the phase transition temperature. The blank LPNs showed peaks at 45.0 °C, 96.7 °C, and 276.4 °C, while oridonin-loaded LPNs showed similar peaks. There was no significant difference between these two samples. The oridonin peak had therefore disappeared in oridonin-loaded LPNs, indicating that the drug may be in an amorphous form. There was no significant difference in the peaks observed for WGA-LPNs, as compared with LPNs, indicating that WGA modification did not influence this property of the nanoparticles.
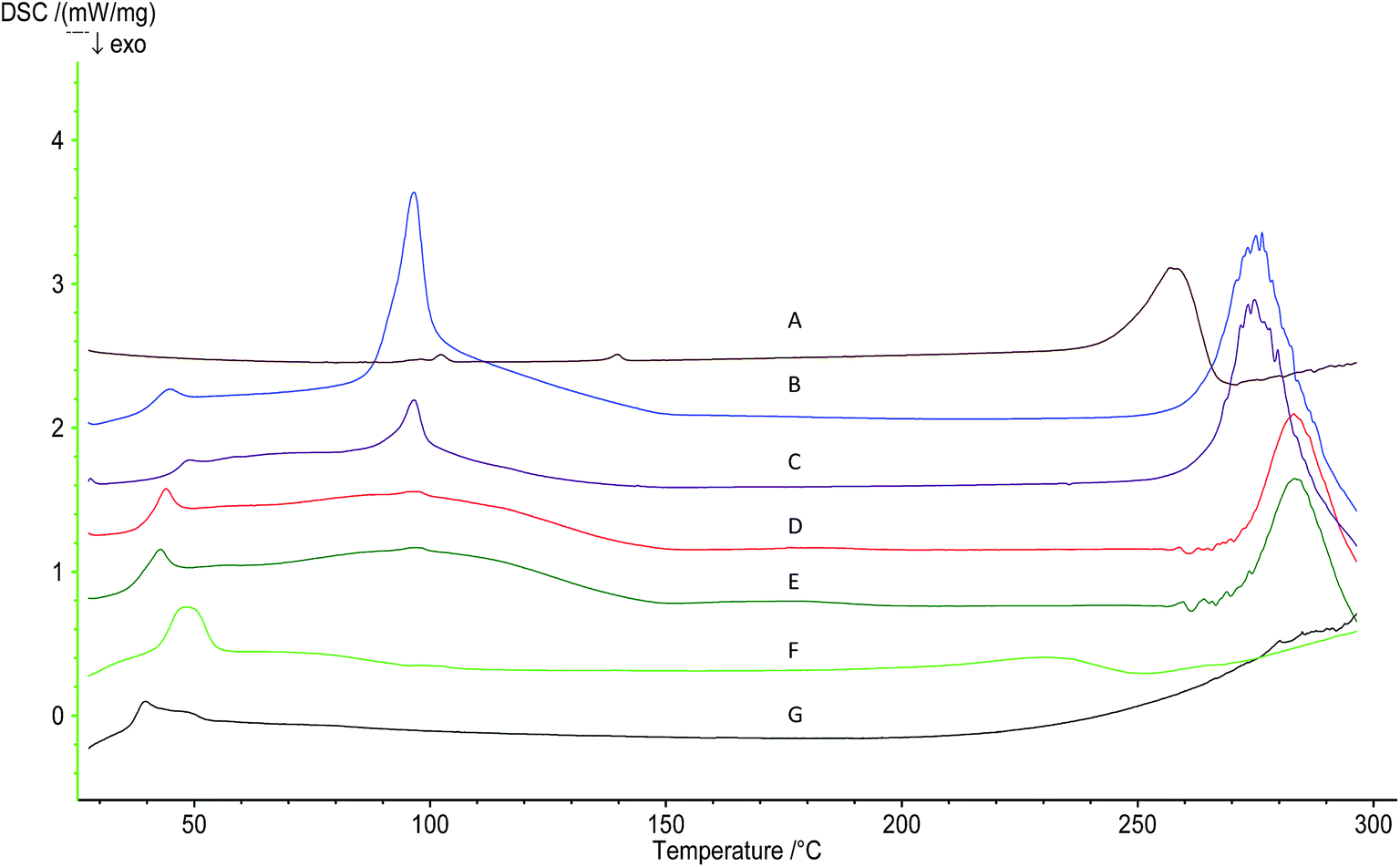 | ||
| Fig. 5 DSC curves of (A) oridonin; (B) blank-LPNs; (C) oridonin-loaded LPNs; (D) blank-WGA-LPNs; (E) oridonin-loaded WGA-LPNs; (F) physical mixture; (G) PLGA. | ||
3.3. In vitro release
A previous study showed that oridonin was slightly unstable in the release medium, with approximately 8% degradation observed over 48 h.42 Therefore, the present study used a modified method that determined the level of oridonin within the dialysis bag, rather than in the release medium. As shown in Fig. 6, burst release of approximately 54% oridonin from the LPNs occurred in the first 2 h, and 87% had been released by 48 h. To investigate the reason for this burst release, the distribution of oridonin in the lipid layer and polymeric core was determined. The percentage of oridonin in the lipid layer was 85.3 ± 5.2%, with 14.7 ± 7.0% located in the polymeric core. This indicated that the release process in the initial period reflected a strong interaction between oridonin in the lipid layer and the release medium, followed by a more gradual release of the remaining lipid-layer oridonin, as well as release from the PLGA core. This release curve showed a biphasic manner, similar with the previous study,43 which was illustrated according to the Gallagher–Corrigan model that the fraction of drug released in the initial burst phase was due to the dissolution of drug at the surface of nanoparticles. According to the f2 factor 65.3 < 100.0, there was no significant difference between LPNs and WGA-LPNs, indicating that WGA modification had no obvious influence on drug release. It should be noted that reducing the rapid release of drugs in the first several hours might be helpful in further improving the bioavailability of oridonin. This may be achieved by altering the distribution of oridonin between the lipid shell and polymeric matrix. | ||
| Fig. 6 In vitro release profiles of oridonin from LPNs and WGA-LPNs. | ||
3.4. Cellular uptake
Enterocytes and goblet cells are the main cells in the gastrointestinal tract. Therefore, this study used Caco-2 cells and HT29-MTX cells to investigate the cellular uptake of LPNs and WGA-LPNs.As shown in Fig. 7A, both LPNs and WGA-LPNs showed time-dependent uptake behaviour, with WGA-LPNs showing lower uptake into both cell-types over the first 0.5 h. This difference had disappeared by 1 h, and WGA-LPNs then showed a higher percentage uptake than LPNs at 2 h and 3 h. This may reflect the different physicochemical features of these particles, such as their size, shape, surface chemistry and ligand modification. These factors have previously been shown to influence the interactions between nanoparticles and the cell membrane.44,45
 | ||
| Fig. 7 Percentage uptake of LPNs and WGA-LPNs by Caco-2 and HT29-MTX cells (mean ± SD, n = 3) (A) at the indicated time-points and (B) in the presence of the indicated inhibitors. *p < 0.05, **p < 0.01. (C) Confocal microscopy showing coumarin 6 (top row, green), Hoechst 33258 (second row, blue) and merged images (third row). | ||
To investigate the cellular uptake mechanism further, we used sodium azide, a metabolic inhibitor that inhibits endocytosis by depletion of cellular ATP. As shown in Fig. 7B, cellular uptake was significantly decreased in the presence of sodium azide (p < 0.05). This result indicated that the nanoparticles were taken up via an energy-dependent process. We performed a competition experiment using N-acetyl-D-glucosamine to investigate whether the increased cellular uptake of WGA-LPNs, as compared with LPNs, was due to the WGA ligand modification. As shown in Fig. 7B, the percentage WGA-LPNs uptake decreased significantly in the presence of N-acetyl-D-glucosamine, suggesting that it competed for the same binding site. Therefore, receptor-mediated endocytosis is one of the factors likely to be involved in the cellular uptake of WGA-LPNs. The presence of N-acetyl-D-glucosamine also reduced cellular LPNs uptake, perhaps due to changes in cell surface area or osmotic gradients in the presence of this compound.46 In addition, we investigated the contribution of clathrin-mediated endocytosis in the uptake of LPNs and WGA-LPNs. In the presence of chlorpromazine, which disrupts clathrin assembly, LPNs and WGA-LPNs uptake was significantly reduced. This indicated that clathrin-mediated endocytosis contributed to the uptake of LPNs and WGA-LPNs. Clathrin-mediated endocytosis has been reported to contribute to the internalisation of many nanoparticles.47,48 Particle size influences cellular uptake and the mechanism underlying endocytosis.49 LPNs were approximately 200 nm, which would be appropriate for clathrin-mediated endocytosis. Clathrin-mediated endocytosis is also involved in the cellular uptake of WGA-LPNs, despite their larger particle size (>300 nm). In some studies, clathrin-mediated endocytosis also occurred with larger particles (>1 μm), suggested that particle size is not the only criterion for particle internalization by this mechanism.50 There are some other endocytosis pathways that may mediate nanoparticle internalisation, including phagocytosis and caveolae-mediated endocytosis. Phagocytosis is considered to be the main internalisation mechanism used by professional phagocytes,51 but this makes less of a contribution in non-professional phagocytes, such as the epithelial cells used in this study. In addition, caveolae-mediated endocytosis occurs for nanoparticles that are 20–100 nm in size. These two processes were thus less likely to play a major role in the cellular uptake observed in this study. However, it should be noted that uptake behaviour and the underlying mechanisms are very complicated and are influenced by both the nanoparticle features and by the cells.52 Further study will be required to elucidate this in full.
In this study, co-cultures of Caco-2 and HT29-MTX cells at a ratio of 9:1 were used to observe cellular uptake of LPNs and WGA-LPNs using confocal laser scanning microscopy. Hoechst 33258, a blue fluorescent dye that binds the minor groove of double stranded DNA, was used to mark the location and morphology of the cell nucleus. As shown in Fig. 7C, uptake of each type of nanoparticle increased over time. WGA-LPNs showed greater fluorescence intensity than LPNs at each incubation time-point, indicating that WGA modification enhanced uptake.
3.5. In vivo bioadhesion
Fig. 8 shows a tissue section image of villi in the ligated intestinal loop model after administration of coumarin 6-loaded LPNs WGA-LPNs. Coumarin 6 signals were visible but weak for the first 0.5 h, indicating that LPNs adhered to the villi and were internalised by the cells to a certain extent. The intracellular LPN level increased overtime. The use of WGA-LPNs produced more intense fluorescent signals at a given time-point, as compared to LPNs. In addition, deep penetration was observed in the villi. This most likely reflects WGA-mediated bioadhesion. WGA has been used to facilitate the oral absorption of some proteins, as well as nanoparticles23,53 and liposomes.19 It shows bioadhesion to the intestinal enterocytes and intestinal mucosa because it binds to N-acetyl-D-glucosamine and sialic acid residues. Therefore, WGA-LPNs could effectively adhere to the intestine for an extended period of time. Further in vivo studies are warranted to determine the extent to which WGA-LPNs can improve the bioavailability of poorly water-soluble drugs. | ||
| Fig. 8 Confocal laser scanning microscopy images of villi in the jejunum after administration of coumarin 6 (green signal)-loaded LPNs or WGA-LPNs for the indicated time-periods. The Hoechst 33258 signal is blue. | ||
4. Conclusions
In this study, a nanocarrier with a specific bioadhesive feature was developed in order to improve intestinal bioadhesion and promote cellular drug uptake. The drug delivery system was developed by surface modification of LPNs with a specific bioadhesive material, WGA, and incorporated a poorly water-soluble drug, oridonin. Immuno gold TEM imaging confirmed the morphology of WGA-LPNs and WGA binding. WGA modification facilitated the interaction of LPNs with both Caco-2 and HT29-MTX cells; this was attributed to receptor-mediated endocytosis. In addition, WGA modification improved bioadhesion in the villi, leading to a significantly higher cellular uptake in the rat intestine. Our results suggested that surface modification of LPNs with WGA provides a promising strategy for oral delivery of poorly water-soluble drugs. In addition to the advantages of LPNs, WGA-LPNs can increase cellular uptake and intestinal bioadhesion, which should improve oral drug delivery.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 81303232, No. 81202925).References
- T. Ikezoe, S. S. Chen, X. J. Tong, D. Heber, H. Taguchi and H. P. Koeffler, Int. J. Oncol., 2003, 23, 1187–1193 CAS.
- S. Wang, Z. Zhong, J. B. Wan, W. Tan, G. Wu, M. Chen and Y. Wang, Am. J. Chin. Med., 2013, 41, 177–196 CrossRef CAS PubMed.
- J. Huang, L. Wu, S. Tashiro, S. Onodera and T. Ikejima, J. Pharmacol. Sci., 2008, 107, 370–379 CrossRef CAS.
- C. Y. Li, E. Q. Wang, Y. Cheng and J. K. Bao, Int. J. Biochem. Cell Biol., 2011, 43, 701–704 CrossRef CAS PubMed.
- W. Tian and S. Y. Chen, Chin. J. Integr. Med., 2013, 19, 315–320 CrossRef CAS PubMed.
- B. A. Owona and H. J. Schluesener, Drugs R&D, 2015, 15, 233–244 CAS.
- D. Li, Q. Cui, S. G. Chen, L. J. Wu, S. Tashiro, S. Onodera and T. Ikejima, J. Pharmacol. Sci., 2007, 105, 22–33 CrossRef CAS.
- K. Osawa, H. Yasuda, T. Maruyama, H. Morita, K. Takeya and H. Itokawa, Phytochemistry, 1994, 36, 1287–1291 CrossRef CAS PubMed.
- A. P. Hu, J. M. Du, J. Y. Li and J. W. Liu, Inflammation Res., 2008, 57, 163–170 CrossRef CAS PubMed.
- P. Zhang, Y. Liu, N. Feng and J. Xu, Int. J. Pharm., 2008, 355, 269–276 CrossRef CAS PubMed.
- X. Zhou, X. Zhang, Y. Ye, T. Zhang, H. Wang, Z. Ma and B. Wu, Int. J. Pharm., 2015, 479, 391–398 CrossRef CAS PubMed.
- D. Zheng, C. Duan, D. Zhang, L. Jia, G. Liu, Y. Liu, F. Wang, C. Li, H. Guo and Q. Zhang, Int. J. Pharm., 2012, 436, 379–386 CrossRef CAS PubMed.
- X. Z. Yang, S. Dou, Y. C. Wang, H. Y. Long, M. H. Xiong, C. Q. Mao, Y. D. Yao and J. Wang, ACS Nano, 2012, 6, 4955–4965 CrossRef CAS PubMed.
- L. Zhang, D. Zhu, X. Dong, H. Sun, C. Song, C. Wang and D. Kong, Int. J. Nanomed., 2015, 10, 2101–2114 CAS.
- Y. Liu, J. Pan and S. S. Feng, Int. J. Pharm., 2010, 395, 243–250 CrossRef CAS PubMed.
- A. L. Palange, D. Di Mascolo, C. Carallo, A. Gnasso and P. Decuzzi, Nanomedicine, 2014, 10, 991–1002 CAS.
- G. Ling, P. Zhang, W. Zhang, J. Sun, X. Meng, Y. Qin, Y. Deng and Z. He, J. Controlled Release, 2010, 148, 241–248 CrossRef CAS PubMed.
- C. Wang, P. C. Ho and L. Y. Lim, Int. J. Pharm., 2010, 400, 201–210 CrossRef CAS PubMed.
- A. Makhlof, S. Fujimoto, Y. Tozuka and H. Takeuchi, Eur. J. Pharm. Biopharm., 2011, 77, 216–224 CrossRef CAS PubMed.
- Y. Liu, P. Wang, C. Sun, J. Zhao, Y. Du, F. Shi and N. Feng, Int. J. Pharm., 2011, 419, 260–265 CrossRef CAS PubMed.
- N. Zhang, Q. Ping, G. Huang, W. Xu, Y. Cheng and X. Han, Int. J. Pharm., 2006, 327, 153–159 CrossRef CAS PubMed.
- G. Ratzinger, X. Wang, M. Wirth and F. Gabor, J. Controlled Release, 2010, 147, 187–192 CrossRef CAS PubMed.
- Y. Yin, D. Chen, M. Qiao, Z. Lu and H. Hu, J. Controlled Release, 2006, 116, 337–345 CrossRef CAS PubMed.
- X. Yang, C. G. Koh, S. Liu, X. Pan, R. Santhanam, B. Yu, Y. Peng, J. Pang, S. Golan, Y. Talmon, Y. Jin, N. Muthusamy, J. C. Byrd, K. K. Chan, L. J. Lee, G. Marcucci and R. J. Lee, Mol. Pharm., 2009, 6, 221–230 CrossRef CAS PubMed.
- D. L. Iden and T. M. Allen, Biochim. Biophys. Acta, 2001, 1513, 207–216 CrossRef CAS.
- J. Tang, L. Zhang, Y. Liu, Q. Zhang, Y. Qin, Y. Yin, W. Yuan, Y. Yang, Y. Xie, Z. Zhang and Q. He, Int. J. Pharm., 2013, 454, 31–40 CrossRef CAS PubMed.
- E. Koren, A. Apte, A. Jani and V. P. Torchilin, J. Controlled Release, 2012, 160, 264–273 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, W. Weecharangsan, L. Piao, M. Darby, Y. Mao, R. Koynova, X. Yang, H. Li, S. Xu, L. J. Lee, Y. Sugimoto, R. W. Brueggemeier and R. J. Lee, Int. J. Pharm., 2010, 390, 234–241 CrossRef CAS PubMed.
- Y. Liu, P. Wang, C. Sun, N. P. Feng, W. Zhou, Y. Yang, R. Tan, Z. Chen, S. Wu and J. H. Zhao, Int. J. Pharm., 2010, 397, 155–163 CrossRef CAS PubMed.
- T. Lesuffleur, A. Barbat, E. Dussaulx and A. Zweibaum, Cancer Res., 1990, 50, 6334–6343 CAS.
- H. Kitamura, M. Cho, B. H. Lee, J. R. Gum, B. B. Siddiki, S. B. Ho, N. W. Toribara, T. Lesuffleur, A. Zweibaum, Y. Kitamura, S. Yonezawa and Y. S. Kim, Eur. J. Cancer, 1996, 32, 1788–1796 CrossRef.
- C. Marianecci, D. Paolino, C. Celia, M. Fresta, M. Carafa and F. Alhaique, J. Controlled Release, 2010, 147, 127–135 CrossRef CAS PubMed.
- X. Gao, W. Tao, W. Lu, Q. Zhang, Y. Zhang, X. Jiang and S. Fu, Biomaterials, 2006, 27, 3482–3490 CrossRef CAS PubMed.
- Y. Jin, Y. Song, X. Zhu, D. Zhou, C. Chen, Z. Zhang and Y. Huang, Biomaterials, 2012, 33, 1573–1582 CrossRef CAS PubMed.
- L. Wrobel and D. Collins, Biochim. Biophys. Acta, 1995, 1235, 296–304 CrossRef.
- J. M. Gamboa and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 800–810 CrossRef CAS PubMed.
- K. Hadinoto, A. Sundaresan and W. S. Cheow, Eur. J. Pharm. Biopharm., 2013, 85, 427–443 CrossRef CAS PubMed.
- J. M. Chan, L. Zhang, K. P. Yuet, G. Liao, J. W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS PubMed.
- P. M. Valencia, P. A. Basto, L. Zhang, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2010, 4, 1671–1679 CrossRef CAS PubMed.
- A. Semalty, M. Semalty and D. Singh, J. Inclusion Phenom. Macrocyclic Chem., 2010, 67, 253–260 CrossRef CAS.
- Y. Li, D. J. Yang, S. L. Chen, S. B. Chen and A. S. C. Chan, Pharm. Res., 2007, 25, 563–577 CrossRef PubMed.
- J. Xu, J. Zhao, J. Wang, N. Feng, R. Tan and Y. Liu, Zhongguo Zhongyao Zazhi, 2009, 34, 47–49 CAS.
- K. M. Gallagher and O. I. Corrigan, Mechanistic aspects of the release of levamisole hydrochloride from biodegradable polymers, J. Controlled Release, 2000, 69, 261–272 CrossRef CAS PubMed.
- C. M. Beddoes, C. P. Case and W. H. Briscoe, Adv. Colloid Interface Sci., 2015, 218, 48–68 CrossRef CAS PubMed.
- H. M. Ding and Y. Q. Ma, Biomaterials, 2012, 33, 5798–5802 CrossRef CAS PubMed.
- P. Arbós, M. Wirth, M. A. Arangoa, F. Gabor and J. M. Irache, J. Controlled Release, 2002, 83, 321–330 CrossRef.
- H. Y. Nam, S. M. Kwon, H. Chung, S. Y. Lee, S. H. Kwon, H. Jeon, Y. Kim, J. H. Park, J. Kim, S. Her, Y. K. Oh, I. C. Kwon, K. Kim and S. Y. Jeong, J. Controlled Release, 2009, 135, 259–267 CrossRef CAS PubMed.
- A. R. Neves, J. F. Queiroz, S. A. Costa Lima, F. Figueiredo, R. Fernandes and S. Reis, J. Colloid Interface Sci., 2016, 463, 258–265 CrossRef CAS PubMed.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- E. Veiga and P. Cossart, Trends Cell Biol., 2006, 16, 499–504 CrossRef CAS PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- Y. Shen, J. Chen, Q. Liu, C. Feng, X. Gao, L. Wang, Q. Zhang and X. Jiang, Int. J. Pharm., 2011, 413, 184–193 CrossRef CAS PubMed.
- B. Y. Kim, J. H. Jeong, K. Park and J. D. Kim, J. Controlled Release, 2005, 102, 525–538 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04023c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |