
Abstract
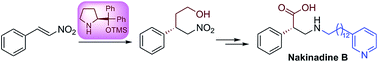
A novel approach for the synthesis of α-phenyl-β2-amino acid core unit 1 and its application to the total synthesis of (S)-nakinadine B 3, a marine natural product, is described. The synthesis utilizes the optimized combination of diphenylprolinol silyl ether mediated asymmetric Michael addition and a proline catalyzed aminoxylation reactions as key steps.
 Please wait while we load your content...
Please wait while we load your content...

