
Expeditious synthesis of the tetrasaccharide cap domain of the Leishmania donovani lipophosphoglycan using one-pot glycosylation reactions†
Mana Mohan Mukherjee,
Nabamita Basu‡
and
Rina Ghosh*
Department of Chemistry, Jadavpur University, Jadavpur, Kolkata 700032, India. E-mail: ghoshrina@yahoo.com
First published on 25th April 2016
Abstract
Expeditious syntheses of the tetrasaccharide cap related to the lipophosphoglycan of Leishmania donovani from two monosaccharides and a disaccharide building block were achieved by one-pot sequential glycosylation reactions. The monosaccharide building blocks were synthesized from D-mannose, and the disaccharide building block was prepared from lactose by C2-epimerisation of its glucose unit through C2 hydroxy oxidation–reduction.
Introduction
Leishmaniasis, a vector-borne parasitic disease caused by obligate intracellular protozoa, is endemic in more than 88 countries affecting over 12 million people worldwide, causing the deaths of 59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people, and leading to almost 1.5 to 2 million new cases each year.1 Unfortunately, there is no effective vaccine for the treatment of leishmaniasis and the approved drugs suffer from a series of disadvantages like complicated side effects, high cost and ineffectiveness to patients residing at many corners of the world.2 The visceral form of leishmaniasis (VL) is also known as kala-azar (Hindi for black sickness or fever) and causes swelling of the liver and spleen. It is often lethal without treatment. Shockingly, about 500000 new cases of VL occur each year, and approximately 50 percent of the reported VL infections occur in the Asian subcontinent.3 Although kala-azar is most threatening in this subcontinent, cases have also been diagnosed in overseas travellers and US Gulf War veterans.4 Unfortunately both human immunodeficiency virus (HIV) and VL exert a synergistic detrimental effect as they target similar immune cells.5
000 people, and leading to almost 1.5 to 2 million new cases each year.1 Unfortunately, there is no effective vaccine for the treatment of leishmaniasis and the approved drugs suffer from a series of disadvantages like complicated side effects, high cost and ineffectiveness to patients residing at many corners of the world.2 The visceral form of leishmaniasis (VL) is also known as kala-azar (Hindi for black sickness or fever) and causes swelling of the liver and spleen. It is often lethal without treatment. Shockingly, about 500000 new cases of VL occur each year, and approximately 50 percent of the reported VL infections occur in the Asian subcontinent.3 Although kala-azar is most threatening in this subcontinent, cases have also been diagnosed in overseas travellers and US Gulf War veterans.4 Unfortunately both human immunodeficiency virus (HIV) and VL exert a synergistic detrimental effect as they target similar immune cells.5
Direct interactions via the carbohydrate sites of lipophosphoglycans (LPG), which are present abundantly on the promastigote, cause receptor mediated phagocytosis by macrophages and binding of the parasite to the epithelial cells of the sandfly midgut.6 It has been reported that LPG is antigenic and also a virulent factor for survival of the parasite and its infectivity.6,7 LPG deficient mutant Leishmania can neither survive in vector sandfly nor transform to their amastigote form but insertion of exogenous LPG into the plasma membrane can restore both of these functions and can cause risky infection.7b,8 An earlier structural analysis of Leishmania donovani LPG9 showed that heterogeneous LPG consists of four distinct domains:10 (i) a neutral oligosaccharide cap at the terminal non reducing end, (ii) a variable phosphoglycan repeating unit, (iii) a conserved phosphosaccharide and (iv) a glycophosphatidylinositol (GPI) anchor. The unique features of the structure include a 1,4-β linkage between galactose and mannose in the cryptic tetrasaccharide cap, and phosphoglycan repeats. Each of the domains has been studied and shown to display important structure–activity relationships.11 The neutral oligosaccharide cap contains a signal for termination of phosphoglycan assembly6 and an epitope for recognition by macrophage receptors.12 Attachment of the parasite to the digestive tract of the sandfly and human macrophages is also mediated by this tetrasaccharide cap.
The role of LPG in host–parasite interactions led to major interest from synthetic organic chemists who worked towards its total synthesis and its evaluation for chemotherapeutic vaccine design. The first synthesis of the tetrasaccharide cap was reported by Fraser-Reid.13 Although thereafter a few other research groups synthesized the cap tetrasaccharide,14–17 the introduction of an economic one-pot total synthesis is still necessary and requires the refinement of the entire synthetic protocol.
In a continuation of our work on oligosaccharide syntheses,18 we report herein the synthesis of the saccharide cap A (Fig. 1), related to the repeating unit present in the LPG of Leishmania donovani, as its 3-(N-benzyloxycarbonyl)propyl glycoside (1, Fig. 2) by sequential one-pot protocols, from the corresponding monosaccharide and disaccharide building blocks.
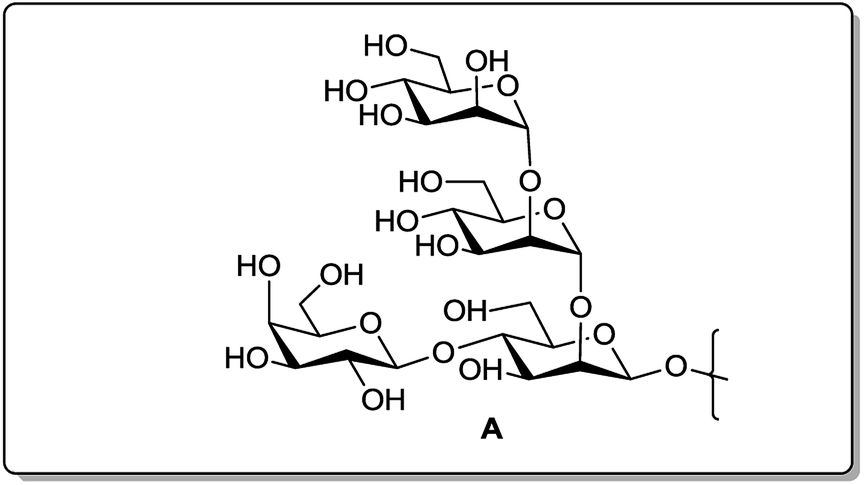 | ||
| Fig. 1 Tetrasaccharide cap domain of Leishmania donovani. | ||
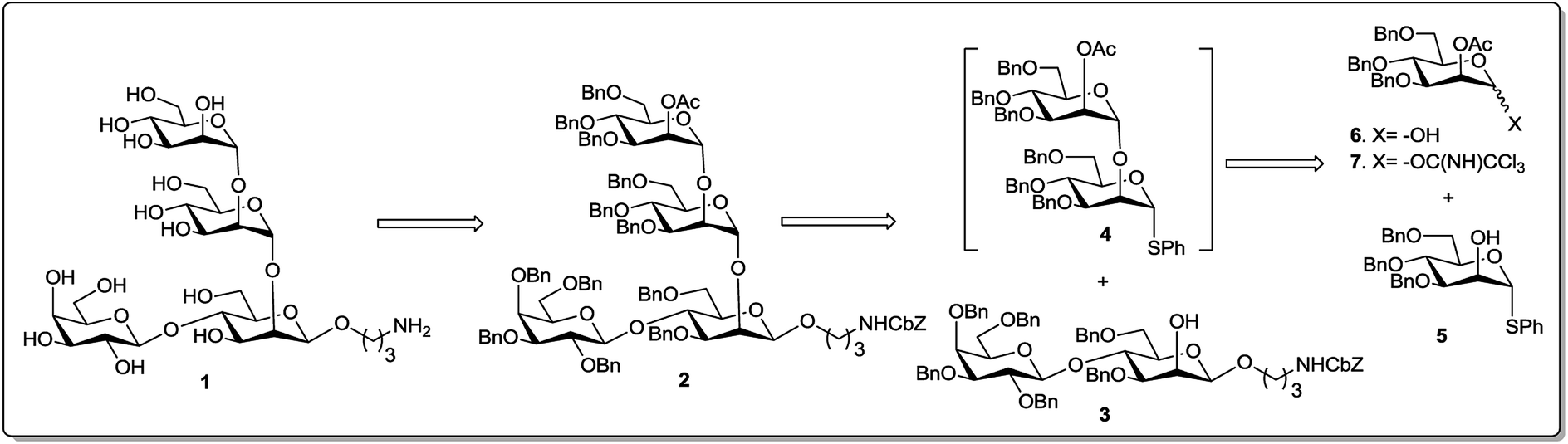 | ||
| Fig. 2 Retro-synthetic analysis of 1. | ||
Result and discussion
For one-pot total synthesis of the tetrasaccharide cap of Leishmania donovani two different pathways have been envisioned. The first requires sequential glycosylation based on a trichloroacetimidate donor and the second is based on a hydroxy donor. For each pathway, retro-synthetic analysis of the fully protected tetrasaccharide 2 led to three building blocks: two orthogonal donors, D-mannose-hydroxy 6 or -trichloroacetimidate 7; thiomannoside 5; and a disaccharide acceptor 3 (Fig. 2). The first glycosyl donor 6 or 7 is a super armed donor with 2-O-acetyl and 3,4,6-tri-O-benzyl protection so that glycosylation through acetoxonium ions produces only α-disaccharides via neighbouring group participation. The second mannose component, phenyl 3,4,6-tri-O-benzyl-1-thio-α-mannopyranoside 5, was chosen to serve a dual purpose during the course of the reaction. At the first step of the sequential glycosylation, it would serve as a glycosyl acceptor with the 2-OH group and then at the second step as a thioglycosyl donor. We thought to prepare the reducing end disaccharide acceptor, 3-(N-benzyloxycarbonyl)propyl glycoside 3, with C-2 axial free OH from lactose (Gal-β 1→4-Glc) via a suitable protecting group manipulation and gluco–manno conversion through inversion of the C-2 centre to give Gal-β 1→4-Man.Thus, phenyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-2-O-acetyl-3,6-di-O-benzyl-1-thio-β-D-glucopyranoside 919 was prepared from commercially available D-lactose via the 1,2-orthoester 819 (Scheme 1). The reported yield for this thiolation of the 1,2-orthoester 8 was very poor at only 22%. After several trials we modified the reaction conditions by increasing the thiophenol equivalency to 5 and decreasing the reaction time to 1.5 hour. These modifications generated the desired product 9 in 78% yield. Glycosylation of thioglycoside 9 with benzyl-N-(3-hydroxy propyl)carbamate using trichloroisocyanuric acid (TCCA) and TMSOTf20 produced 3-(N-benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-2-O-acetyl-3,6-di-O-benzyl-β-D-glucopyranoside 10 in 96% yield. Zemplén deacetylation21 of compound 10 furnished 3-(N-benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-β-D-glucopyranoside 11 in almost quantitative yield. To prepare the projected acceptor we needed to invert the equatorial C2 hydroxy group of 11. Initially its conversion to the corresponding triflate followed by an SN2 inversion at the C2 was envisioned by us, but this sequence of reactions did not result in the desired outcome. Thus we opted for an alternate route viz., C2 oxidation-stereoselective reduction,17,22 to achieve the desired galactose-(1→4)-mannose acceptor. With this end in view, compound 11 was oxidised using Dess–Martin periodinane (DMPI)23 in dry CH2Cl2 in the presence of catalytic pyridine to produce the corresponding 2-keto compound. Direct reduction of that ketone with sodium borohydride in methanol produced the desired acceptor 3-(N-benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-β-D-mannopyranoside 3. The change in substrate polarity (checked by TLC) and spectral analysis confirmed this inversion via oxidation–reduction with 76% yield.
 | ||
| Scheme 1 Synthesis of the disaccharide acceptor 3. Reagents and conditions: (a) I2, Ac2O, l h, quantitative. (b) AcOH, 33% HBr–AcOH, 5 h. (c) 2,6-Leutidine, MeOH:DCM 3:1, TBAB, 72 h, 81%. (d) BnCl, KOH, THF, 60 °C, 8 h, 88%. (e) PhSH, HgBr2, MeCN, 60 °C, 1.5 h, 78%. (f) OH(CH2)3NHCbZ, TCCA, TMSOTf, 0 °C, DCM, 10 min, 96%. (g) NaOMe, MeOH:DCM (1:1), 3 h, quantitative. (h), (i) DMPI, DCM, Py, 2 h. (ii) NaBH4, MeOH, overnight, 76% over 2 steps. | ||
The carbohydrate moiety 1424 was prepared via the 1,2-orthoester 1324a,b of commercially available D-mannose using reported methods (Scheme 2). Zemplén deacetylation21 of compound 14 furnished phenyl 3,4,6-tri-O-benzyl-1-thio-D-mannopyranoside 524a in quantitative yield. Thioglycoside hydrolysis25 of compound 14 was then effected using TCCA in wet acetone to afford 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-D-mannopyranose 6 in 96% yield. Treatment of 6 with trichloroacetonitrile and DBU in dry CH2Cl2 produced the acetimidate donor 724b in 91% yield.
 | ||
| Scheme 2 Synthesis of D-mannose based donors and acceptor. Reagents and conditions: (a) Ac2O, I2, rt, 15 min, quantitative. (b) AcOH, 33% HBr–AcOH, 5 h. (c) 2,6-Leutidine, DCM:MeOH 3:1, 8 h, 92%. (d) BnCl, KOH, 60 °C, 8 h, 87%. (e) PhSH, HgBr2, MeCN, 60 °C, 2 h, 92%. (f) NaOMe, MeOH, rt, 3 h, quantitative. (g) TCCA, (CH3)2CO–H2O, 30 min, 96%. (h) CCl3CN, DBU, CH2Cl2, 91%. | ||
After being equipped with the appropriately tailored building blocks, the one-pot sequential glycosylation reactions leading to the protected tetrasaccharide 2 were started from the reducing end of the compound. Trichloroacetimidate donor 7 and thioglycoside acceptor 5 were allowed to couple using 20 mol% TMSOTf26 in dry CH2Cl2 at 0 °C (Scheme 3). After 20 minutes, the consumption of both of the starting materials (as indicated by TLC) resulted in a clean conversion to the desired disaccharide 4. Into the same reaction vessel, the non reducing acceptor 3 was added along with 1 equivalent of NIS. The reaction temperature was cooled to −30 °C and again 20 mol% TMSOTf was added to it. After 30 minutes, TLC showed full conversion to the fully protected tetrasaccharide, and the reaction was quenched by the addition of NEt3 at that temperature. The crude product was purified by column chromatography providing an 87% yield of the pure desired tetrasaccharide derivative 2.
 | ||
| Scheme 3 Sequential one-pot synthesis of the tetrasaccharide derivative 2. | ||
After achieving the final protected tetrasaccharide via trichloroacetimidate donor activation, we tried to rebuild the same tetrasaccharide in a more economical way, i.e. via sequential one-pot glycosylation based on the initial 1-hydroxy donor.27 For that purpose, to a mixture of 1-hydroxy donor 6, Ph2SO and TTBP in CH2Cl2 at −60 °C was added Tf2O. The temperature was raised to −40 °C and maintained for 1 hour. Then acceptor 5 was added, and the reaction mixture was allowed to reach room temperature slowly during 3 hours. After a clean conversion to the disaccharide (as indicated by TLC) the reaction mixture was again cooled to −60 °C, and additional Tf2O was added. After 10 minutes at that temperature, acceptor 3 was added, and the resulting mixture was allowed to reach 0 °C. TLC was used to check the reaction's progress after 1 hour, and the reaction was quenched by addition of NEt3. The crude product was purified by column chromatography giving an 82% yield of the pure tetrasaccharide derivative 2; this yield was comparable with that based on the previous approach.
NMR and HRMS analyses of compound analyses of compound 2 unambiguously confirmed its formation and structure. Finally deacetylation under Zemplén conditions, followed by global debenzylation using hydrogen and palladium-charcoal in a mixture of water and methanol, afforded the desired tetrasaccharide 1 (Scheme 4). Compound 1 was characterised by NMR techniques (1H-, 13C-, COSY and HSQC) and also by HRMS. The anomeric protons of compound 1 appear at δ 5.44 (b, H1′′′), 4.87 (bs, H1), 4.76 (bs, H1′′) and 4.44 (d, J 8 Hz, H1′) ppm and the corresponding carbons appear at 99.9, 100.8, 100.5 and 103.1 ppm, respectively.
 | ||
| Scheme 4 Deprotection of the tetrasaccharide derivative 2. | ||
Conclusion
In conclusion, the syntheses of the tetrasaccharide cap of Leishmania donovani via one-pot sequential glycosylation techniques were successfully achieved in excellent yields. A number of notable features are: (a) inversion at C2 of the D-glucosyl moiety of lactose by applying an oxidation-stereoselective reduction approach; (b) step-by-step economic synthesis of the glycosyl donors and acceptors as well as the glycosylated products; and (c) [1 + 1 + 2] one-pot sequential glycosylations. All the reactions described are clean, stereoselective and high yielding.Experimental
NMR spectra were recorded on a Bruker DPX 300 NMR spectrometer operating at 300 and 500 MHz for 1H-NMR, and at 75, 100 and 125 MHz for 13C-NMR in CDCl3 and D2O. HRMS data were recorded on a Q-tof-Micro mass spectrometer using the electron spray ionization method. Specific rotations were measured on a Jasco J-815 spectrometer.Phenyl 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside (14)24
To a solution of 3,4,6-tri-O-benzyl-β-D-mannopyranosyl-1,2-(methyl orthoacetate) (13, 2.88 g, 5.70 mmol), in dry CH3CN (30 mL) containing 4 g of molecular sieves (4 Å), thiophenol (5.5 mL, 28.5 mmol, 5 equiv.) and HgBr2 (205 mg, 0.57 mmol, 0.1 equiv.) were added at room temperature. This mixture was heated with stirring at 60 °C under an argon atmosphere for 2 hours, and then cooled to room temperature. The reaction mixture was filtered through a Celite bed, and the residue was washed with CH2Cl2 (3 × 5 mL). The combined filtrate and washings were washed subsequently with cold 5% aqueous NaOH (250 mL) followed by water (2 × 100 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel (PE/EtOAc, 9:1) to afford 14 as a white solid (3.02 g, 92%); m.p. 64–66 °C; [α]24D +105.0 (c 1.2, CHCl3); lit24 m.p. 62–63 °C; [α]19D +106.0 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 2.16 (s, 3H, COCH3), 3.73 (dd, 1H, J = 1.7, 10.8 Hz), 3.87 (dd, 1H, J = 4.4, 10.8 Hz), 3.93–4.02 (m, 2H), 4.34 (m, 1H), 4.46–4.60 (m, 3H), 4.69 (d, 1H, J = 19.6 Hz), 4.73 (d, 1H, J = 18.8 Hz), 4.90 (d, 1H, J = 10.7 Hz), 5.55 (s, 1H), 5.62 (d, 1H, J = 1.5 Hz), 7.19–7.36 (m, 18H, ArH), 7.47–7.50 (m, 2H, ArH). 13C NMR (75 MHz, CDCl3): δ 21.1, 68.9, 70.4, 72.0, 72.5, 73.4, 74.6, 75.3, 78.5, 86.3, 126.6, 127.0, 127.6, 127.69, 127.71, 127.8, 127.9, 128.2, 128.3, 128.4, 128.5, 129.1, 131.9, 133.5, 133.7, 137.6, 138.2, 138.3, 170.3. HRMS (TOF): calc. for (M + Na)+ C35H36O6SNa 607.2131, found 607.2128.
Phenyl 3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside (5)24a
To a solution of 14 (1.0 g, 1.6 mmol) in dry methanol (10 mL), 1 M methanolic NaOMe (0.2 mL) solution was added, and the reaction mixture was stirred for 3 hours at room temperature. The reaction mixture was then neutralized with Dowex-50W cation exchange resin (H+) and filtered. The resin was washed with methanol, and the combined filtrate and washings were concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (PE/EtOAc, 5:1) to give glassy syrupy product 5 (0.92 g, 99%); [α]27D +135.9 (c 2.52, CHCl3); lit24 [αD] +182.5 (c 1.26, CHCl3); 1H NMR (300 MHz, CDCl3): δ 3.69 (m, 1H), 3.81 (m, 1H), 3.86–3.98 (m, 2H), 4.26–4.33 (m, 2H), 4.47 (d, 1H, J = 12.0 Hz), 4.54 (d, 1H, J = 10.8 Hz), 4.63 (d, 1H, J = 12.0 Hz), 4.73 (s, 2H), 4.85 (d, 1H, J = 10.8 Hz), 5.62 (s, 1H), 7.22–7.38 (m, 18H, ArH), 7.46–7.48 (m, 2H, ArH). 13C NMR (75 MHz, CDCl3): δ 68.9, 70.0, 72.2, 72.4, 73.5, 74.6, 75.3, 77.3, 80.4, 87.5, 127.5, 127.6, 127.8, 127.9, 128.0, 128.08, 128.14, 126.17, 128.3, 128.4, 128.5, 128.7, 129.1, 131.7, 133.9, 137.7, 138.3, 138.4. HRMS (TOF): calc. for (M + Na)+ C35H34O4SNa 565.2025, found 565.2024.
2-O-Acetyl-3,4,6-tri-O-benzyl-1-thio-D-mannopyranose (6)24b
A suspension of 14 (1.0 g, 1.6 mmol) and TCCA (372 mg, 1 equiv.) in (CH3)2CO–H2O (4:1) was stirred at 0 °C for 30 minutes. After completion of the reaction (as indicated by TLC) (CH3)2CO was evaporated in vacuo, and the resulting mixture was diluted with CH2Cl2 (15 mL). The solution was washed subsequently with saturated aqueous NaHCO3 (200 mL) and water. The organic layer was dried over anhydrous Na2SO4 and concentrated. The residue was filtered through a column of silica gel (eluent: PE/EtOAc, 4:1) to afford 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-D-mannopyranose 6 as a white foam (808 mg, 96%). 1H NMR (300 MHz, CDCl3): δ 2.16 (s, 3H, COCH3), 3.59–3.78 (m, 5H), 4.03–4.11 (m, 2H), 4.47–4.66 (m, 4H), 4.73 (m, 1H), 4.86 (m, 1H), 5.22 (t, 1H, J = 1.5 Hz), 5.38 (m, 1H), 7.15–7.18 (m, 2H, ArH), 7.27–7.37 (m, 15H, ArH). HRMS (TOF): calc. for (M + Na)+ C29H32O7Na 515.2046, found 515.2040.
2-O-Acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl trichloroacetimidate (7)24b
To a suspension of 6 (800 mg, 1.5 mmol) in dry CH2Cl2 (10 mL), trichloroacetonitrile (0.23 mL, 2.25 mmol, 1 equiv.) and DBU (0.04 mL, 0.45 mmol, 0.3 equiv.) were added dropwise at −5 °C. After completion of the reaction, CH2Cl2 was removed under rotary evaporation. The sticky brown mass was purified by chromatography on a 60–120 mesh silica gel column (PE/EtOAc, 9:1) to give pure compound 7 as a colourless syrup (920 mg, 91%). [α]24D +30.1 (c 1.2, CHCl3); lit24b [α]19D +36.3 (c 0.9, CHCl3); 1H NMR (300 MHz, CDCl3): δ 2.20 (s, 3H, COCH3), 3.72 (m, 1H), 3.85 (m, 1H), 4.00–4.06 (m, 3H), 4.51 (d, 1H, J = 8.1 Hz), 4.54 (d, 1H, J = 6.6 Hz), 4.58 (d, 1H, J = 11.2 Hz), 4.69 (d, 1H, J = 12.1 Hz), 4.74 (d, 1H, J = 11.2 Hz), 4.88 (d, 1H, J = 10.6 Hz), 5.5 (m, 1H), 6.31 (d, 1H, J = 1.7 Hz), 7.17–7.20 (m, 2H, ArH), 7.29–7.37 (m, 13H, ArH), 8.69 (s, NH). HRMS (TOF): calc. for (M + Na)+ C31H32Cl3NO7Na 659.1142, found 659.1148.
Phenyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-2-O-acetyl-3,6-di-O-benzyl-1-thio-β-D-glucopyranoside (9)20
To a solution of 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-α-D-glucopyranose-1,2-(methylorthoacetate) (8, 2.2 g, 2.34 mmol), in dry CH3CN (30 mL) containing 4 g of molecular sieves (4 Å), thiophenol (1.15 mL, 11.7 mmol, 5 equiv.) and HgBr2 (84 mg, 0.23 mmol, 0.1 equiv.) were added at room temperature. This mixture was heated under stirring at 60 °C under an argon atmosphere for 1.5 hours, and then cooled to room temperature. The reaction mixture was filtered through a Celite bed, and the residue was washed with CH2Cl2 (3 × 5 mL). The combined filtrate and washings were subsequently with cold 5% aqueous NaOH (250 mL) followed by water (2 × 100 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel (PE/EtOAc, 9:1) to afford 9 as a white solid. It was crystallized from methanol (1.85 g, 78%); m.p. 94–96 °C; [α]24D −5.9 (c 1.1, CHCl3); lit20 m.p. 96 °C [EtOH]; [α]25D −5.1 (c 0.7, CHCl3); 1H NMR (300 MHz, CDCl3): δ 2.05 (s, 3H, COCH3), 3.33–3.52 (m, 5H), 3.63 (t, 1H, J = 8.9 Hz), 3.78–3.83 (m, 3H), 3.94–4.01 (m, 2H), 4.22–4.35 (m 2H), 4.39–4.49 (m 3H), 4.53–4.67 (m, 3H), 4.74–4.79 (m, 2H), 4.83–4.88 (m, 2H), 4.97–5.07 (m, 3H), 7.22–7.36 (m, 33H, ArH), 7.52–7.55 (m, 2H, ArH). 13C NMR (75 MHz, CDCl3): δ 21.1, 68.3, 68.4, 71.4, 72.7, 73.2, 73.5, 73.7, 74.7, 75.4, 79.8, 80.0, 82.3, 82.6, 86.0 (C1), 103.0 (C1′), 127.05, 127.2, 127.42, 127.48, 127.52, 127.58, 127.6, 127.7, 127.8, 127.9, 128.1, 128.2, 128.3, 128.4, 128.5, 128.9, 132.3, 133.2, 138.47, 138.51, 138.8, 138.9, 139.1, 169.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). HRMS (TOF): calc. for (M + Na)+ C62H64O11SNa 1039.4067, found 1039.4068.
O). HRMS (TOF): calc. for (M + Na)+ C62H64O11SNa 1039.4067, found 1039.4068.
3-(N-Benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-2-O-acetyl-3,6-di-O-benzyl-β-D-glucopyranoside (10)
To a mixture of phenyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-2-O-acetyl-3,6-di-O-benzyl-1-thio-β-D-glucopyranoside 9 (1.8 g, 1.77 mmol) and benzyl-N-(3-hydroxy propyl)carbamate (445 mg, 2.13 mmol, 1.2 equiv.) in dry CH2Cl2 (25 mL), 3 g of flame activated molecular sieves (4 Å) were added. The mixture was stirred at room temperature under an argon atmosphere. After 40 minutes the mixture was cooled to 0 °C, and TCCA (410 mg, 1.77 mmol, 1 equiv.) was added to it. Then TMSOTf (0.096 mL, 0.53 mmol, 0.3 equiv.) was added via a micro-syringe. After 10 minutes, the acceptor was consumed completely (as checked by TLC), the reaction mixture was filtered off through a Celite bed, and the bed was washed with CH2Cl2. The combined filtrate and washings were washed subsequently with a saturated NaHCO3 solution and water. The organic layer was dried over anhydrous Na2SO4 and concentrated to afford the glycosylated product. The crude mass was purified by silica gel column chromatography (60–120 mesh) (PE/EtOAc 3:1) to get pure product 10 (1.89 g) as a colourless syrup in 96% yield. [α]29D −2.57 (c 7.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.68–1.76 (bs, 2H), 1.97 (s, 3H, COCH3), 3.24 (m, 1H), 3.33–3.37 (m, 3H), 3.38–3.43 (m, 2H), 3.46 (m, 1H), 3.55–3.59 (m, 2H), 3.69 (d, 1H, J = 10.0 Hz), 3.74–3.77 (m, 2H), 3.85 (m, 1H), 3.9 (d, 1H, J = 2.5 Hz), 3.95 (t, 1H, J = 9.0 Hz), 4.23 (d, 1H, J = 12.0 Hz), 4.29–4.34 (apparent t, 2H, J = 10.5, 11.5 Hz), 4.36–4.38 (dd, 2H, J = 1.5, 9 Hz), 4.48 (d, 1H, J = 12.0 Hz), 4.53–4.59 (dd, 2H, J = 11.5, 19.0 Hz), 4.67–4.77 (m, 3H), 4.82 (d, 1H, J = 11.0 Hz), 4.94–4.98 (m, 3H), 5.08 (bs, 2H), 5.29 (bs, 1H), 7.19–7.36 (m, 35H, ArH). 13C NMR (75 MHz, CDCl3): δ 20.9, 29.5, 38.2, 66.5, 66.7, 68.1, 68.3, 72.5, 72.6, 73.1, 73.5, 73.7, 74.3, 74.7, 75.3, 79.9, 80.8, 82.5, 100.8 (C1), 102.9 (C1′), 127.2, 127.4, 127.5, 127.57, 127.62, 127.7, 127.9, 128.0, 128.1, 128.2, 128.30, 128.33, 128.4, 128.5, 136.9, 138.1, 138.2, 138.5, 138.8, 139.0, 139.1, 156.6 (CO), 169.6 (CO). HRMS (TOF): calc. for (M + Na)+ C67H73NO14Na 1138.4929, found 1138.4932.
3-(N-Benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-β-D-glucopyranoside (11)
To a solution of 10 (1.85 g, 1.66 mmol) in a mixture of dry CH2Cl2 (10 mL) and dry methanol (8 mL) 1 M methanolic NaOMe (2 mL) solution was added, and the reaction mixture was stirred for 3 hours at room temperature. The reaction mixture was then neutralized with Dowex-50W cation exchange resin (H+) and filtered. The resin was washed with methanol, and the combined filtrate and washings were concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (PE/EtOAc, 3:1) to give glassy syrupy product 11 (1.75 g, 99%); [α]27D −1.14 (c 5.1, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.79–1.81 (bs, 2H), 3.26 (m, 1H), 3.34–3.43 (m, 4H), 3.45–3.49 (m, 2H), 3.55 (t, 1H, J = 8.5 Hz), 3.64–3.69 (m, 2H), 3.74–3.78 (m, 2H), 3.89–3.96 (m, 3H), 4.27–4.32 (m, 3H), 4.36–4.41 (m, 2H), 4.48 (d, 1H, J = 12.0 Hz), 4.55 (d, 1H, J = 11.5 Hz), 4.68–4.71 (m, 2H), 4.75 (m, 2H), 4.81 (d, 1H, J = 11.0 Hz), 4.96 (d, 1H, J = 11.0 Hz), 5.06–5.08 (m, 3H), 5.51 (bs, 1H), 7.17–7.35 (m, 35H). 13C NMR (75 MHz, CDCl3): δ 29.4, 38.2, 66.6, 67.4, 68.3, 72.6, 73.1, 73.5, 73.6, 74.7, 75.27, 75.32, 76.3, 79.9, 82.5, 82.8, 102.7 (C1), 102.8 (C1′), 127.37, 127.48, 127.5, 127.71, 127.75, 127.75, 127.9, 128.0, 128.1, 128.2, 128.27, 128.3, 128.40, 128.42, 128.49, 138.5, 136.8, 138.1, 138.2, 138.8, 139.0, 156.7 (CO). HRMS (TOF): calc. for (M + Na)+ C65H71NO13Na 1096.4823, found 1096.4825.
3-(N-Benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-β-D-mannopyranoside (3)
To a solution of 11 (1.70 g, 1.58 mmol) in a mixture of anhydrous CH2Cl2 (10 mL) and dry pyridine (0.25 mL, 3.16 mmol, 2 equiv.) was added DMPI (1.4 g, 3.16 mmol, 2 equiv.), and the reaction mixture was allowed to stir at room temperature for 2 hours. The reaction mixture was diluted with CH2Cl2 (75 mL), and the organic layer was successively washed with saturated NaHCO3 solution and water, then dried over anhydrous Na2SO4 and finally concentrated under reduced pressure. To the solution of this crude keto product in distilled methanol (30 mL), was added NaBH4 (895 mg, 23.7 mmol, 15 equiv.) and the reaction mixture was allowed to stir overnight at room temperature. The solvent was removed under reduced pressure, and the crude mass was dissolved in CH2Cl2 (100 mL). The organic layer was successively washed with saturated NaHCO3 solution and water, dried over anhydrous Na2SO4 and concentrated under vacuum. The crude mass was purified by silica gel column chromatography (60–120 mesh) (PE/EtOAc 3:2) to get pure product 3 (1.29 g) as a colourless syrup in 76% yield. [α]28D −6.54 (c 2.8, CHCl3); 1H (500 MHz, CDCl3): δ 1.79 (bs, 2H, CH2), 3.29–3.32 (m, 2H), 3.39–3.43 (m, 2H), 3.45–3.48 (m, 2H), 3.53–3.63 (m, 3H), 3.69–3.72 (m, 2H), 3.76 (d, 1H, J = 9.5 Hz), 3.89 (bs, 2H), 4.02–4.05 (m, 2H), 4.29–4.32 (m, 2H), 4.36–4.43 (m, 4H), 4.57 (d, 1H, J = 11.5 Hz), 4.65–4.71 (m, 4H), 4.74–4.78 (m, 2H), 4.94 (d, 1H, J = 11.5 Hz), 5.06–5.09 (bs, 2H), 5.44 (bs, 1H), 7.17–7.31 (m 35H, ArH). 13C NMR (75 MHz, CDCl3): δ 29.6, 38.3, 66.5, 67.3, 68.5, 68.9, 72.3, 72.7, 73.1, 73.5, 74.4, 74.6, 75.0, 75.1, 77.3, 79.1, 79.8, 82.6, 99.8 (C1), 103.2 (C1′), 127.4, 127.5, 127.6, 127.7, 127.8, 127.9, 128.0, 128.1, 128.18, 128.24, 128.3, 128.38, 128.42, 128.5, 136.8, 137.0, 138.2, 138.4, 138.7, 138.9, 156.6 (CO). HRMS (TOF): calc. for (M + Na)+ C65H71NO13Na 1096.4823, found 1096.4826.
3-(N-Benzyloxycarbonyl)propyl (2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→2)-(3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→2)-[(2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl)-(1→4)]-3,6-di-O-benzyl-β-D-mannopyranoside (2)
To a solution of 7 (175.0 mg, 0.26 mmol) and 5 (136.6 mg, 0.24 mmol) in dry CH2Cl2 (60 mL), 8 g of activated molecular sieves (4 Å) were added, and the reaction mixture was stirred under an argon atmosphere for 45 minutes. Then the reaction vessel was placed in a 0 °C cold bath, and TMSOTf (10 μL, 0.05 mmol, 0.2 equiv.) was added to it via a micro-syringe. After 10 min, complete consumption of both the starting materials was observed. Acceptor 3 (223.6 mg, 0.21 mmol) and NIS (58.5 mg, 0.26 mmol, 1 equiv.) were then added to the same vessel. After the addition, the reaction temperature was cooled to −30 °C and another 20 mol% of TMSOTf (10 μL, 0.02 mmol) was added to it. The second step of the reaction was completed after 30 minutes (as indicated by TLC). The reaction mixture was quenched by Et3N, filtered through a Celite bed, and the bed was washed with CH2Cl2 (3 × 15 mL). The combined filtrate and washings were washed subsequently with saturated sodium thiosulphate, aqueous NaHCO3 (2 × 50 mL) and water (2 × 50 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated to furnish a syrupy compound. The crude product was purified by flash column chromatography (eluent: PE/EtOAc, 3:1) to afford the desired fully protected tetrasaccharide 2 as a white foam (359.15 mg, 87%); [α]25D25 −14.93 (c 1.97, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.75 (bs, 2H, CH2), 2.07 (s, 3H, COCH3), 3.11–3.20 (m, 3H, NCH, H3′′, H5′), 3.34–3.38 (m, 3H, NCH, H6, H6′′), 3.47 (apparent t, 1H, J = 7.0, 8.0 Hz, H5′′), 3.55–3.67 (m, 8H, OCH, H2′, H5, H6, H6′, H6′′, H6′′′), 3.75–3.81 (m, 5H, OCH, H2′′, H4′′, H3′, H6′), 3.91–3.98 (m, 2H, H4′′′, H6′′′), 4.20–4.28 (m, 7H, H4, H3′′, 5BnH), 4.33–4.53 (m, 12H, H3, H5′′′, H1′, H1′′, 8BnH), 4.57–4.66 (m, 5H, H2, 4BnH), 4.74–4.80 (apparent t, 2H, J = 13.0 Hz, 2BnH), 4.84–4.90 (apparent t, 3H, J = 12.5, 13.5 Hz, H1, 2BnH), 5.00–5.10 (2d, 3H, J = 11.5, 12.0 Hz, 3BnH), 5.46 (s, 1H, H1′′′), 5.54 (bs, 1H, NH), 5.66 (bs, 1H, H2′′′), 7.07–7.11 (m, 12H, ArH), 7.15–7.32 (m, 51H, ArH), 7.55 (d, 2H, J = 7.0 Hz, ArH). 13C NMR (125 MHz, CDCl3): δ 21.2 (CH3), 29.9 (CH2), 37.8 (NCH2), 66.4 (BnC), 66.6 (C6/C6′), 66.9 (BnC), 68.5 (C6′′), 69.2 (C2′′′), 69.6 (BnC), 70.1(OCH2), 70.3 (C6/C6′), 70.5 (C6′′′), 71.5 (BnC), 71.8 (BnC), 72.4 (BnC), 72.44, 72.6 (BnC), 72.7, 73.2 (BnC), 73.3 (BnC), 74.3 (C4′′′), 74.5 (BnC), 74.6 (BnC), 74.8, 74.9 (BnC), 75.0, 75.1, 75.3, 75.4 (C5′′′), 75.7, 75.9, 78.7, 79.9, 82.7 (C3′′), 83.0 (C5′), 83.4, 97.8 (C1′′′), 99.9 (C1), 101.2 (C1′′), 102.8 (C1′), 127.26, 127.33, 127.4, 127.5, 127.63, 127.66, 127.7, 127.81, 127.86, 127.9, 128.1, 128.2, 128.3, 128.4, 128.5, 128.6, 137.0, 138.6, 139.2, 156.9 (CO), 170.6 (CO). HRMS (TOF): calc. for (M + Na)+ C121H1291NO24Na 2002.8803, found 2002.8810.
3-(N-Benzyloxycarbonyl)propyl (2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→2)-(3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→2)-[(2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl)-(1→4)]-3,6-di-O-benzyl-β-D-mannopyranoside (2)
2-O-Acetyl-3,4,6-tri-O-benzyl-1-thio-D-mannopyranose 6 (74.4 mg, 0.152 mmol), Ph2SO (67.8 mg, 0.335 mmol, 2.2 equiv.), and TTBP (56.8 mg, 0.228 mmol, 1.5 equiv.) were dissolved in CH2Cl2 (20 mL), and the reaction was cooled to −60 °C. Tf2O (0.03 mL, 0.183 mmol, 1.2 equiv.) was added, and the reaction mixture was slowly brought to −40 °C in 1 h, after which phenyl 3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside 5 (74.4 mg, 0.137 mmol, 0.9 equiv.) in 1 mL dry CH2Cl2 was added. The reaction mixture was slowly warmed to room temperature. After 1 hour at room temperature the mixture was cooled to −60 °C, and Tf2O (0.03 mL, 0.183 mmol, 1.2 equiv.) was added. After the reaction had been kept at −60 °C for 10 minutes, 3-(N-benzyloxycarbonyl)propyl 2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-β-D-mannopyranoside 3 (134 mg, 0.122 mmol, 0.8 equiv.) was added. The reaction mixture was warmed to 0 °C and stirred for 1 hour, after which it was quenched with by Et3N, filtered through a Celite bed, and the bed was washed with CH2Cl2 (3 × 15 mL). The combined filtrate and washings were washed subsequently with saturated aqueous NaHCO3 (3 × 50 mL) and water (3 × 50 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated to furnish a syrupy compound. The crude product was purified by flash column chromatography (eluent: PE/EtOAc, 3:1) to afford the desired fully protected tetrasaccharide 2 as a white foam (198.17 mg, 82%); the spectral data matched with that for the substrate synthesized previously.
3-(N-Benzyloxycarbonyl)propyl α-D-mannopyranosyl-(1→2)-(α-D-mannopyranosyl)-(1→2)-[(β-D-galactopyranosyl)-(1→4)]-β-D-mannopyranoside (1)
Protected tetrasaccharide 2 (75 mg, 0.038 mmol) was dissolved in CH2Cl2 and methanol (1:1; 8 mL), and NaOMe (1 M in methanol, 0.5 mL) was added. The mixture was stirred for 2 hours and then the reaction was quenched with Dowex-50W cation exchange resin (H+). The resin was filtered off and then washed with methanol (4 × 5 mL). The combined filtrate and washings were evaporated under reduced pressure. A mixture of the resulting mass and 10% Pd–C (70 mg) was taken in methanol (3 mL) and H2O (1 mL) and stirring was continued under an H2 atmosphere for 24 h. The catalyst was filtered through a Celite bed, and the bed was washed with methanol (3 × 5 mL). The combined filtrate and washings were concentrated under reduced pressure. The product was passed through a 0.45 μm Millipore membrane, and lyophilized to afford 1 as a white foam (22.89 mg, 84%); 1H NMR (500 MHz, D2O): δ 2.03 (m, 2H, CH2), 3.13–3.16 (t, 2H, J = 7.0 Hz, NCH2), 3.38–3.45 (m, 2H), 3.54–3.62 (m, 2H, H4, H2′), 3.65–3.69 (m, 4H, H3′), 3.73–3.86 (m, 10H, OCH, H5, H3′′), 3.94–4.01 (m, 5H, OCH, H4′), 4.05–4.13 (m, 3H, H3, H2′′), 4.31 (d, 1H, J = 2.5 Hz, H2), 4.36 (s, 1H H2′′), 4.44 (d, 1H, J = 8.0 Hz, H1′), 4.76 (s, 1H, H1′′), 4.88 (s, 1H, H1), 5.44 (s, 1H, H1′′′). 13C NMR (125 MHz, D2O): 27.0 (CH2), 37.3 (NCH2), 60.8, 61.3, 61.8, 66.6, 66.9 (OCH2), 67.2, 68.8, 70.2, 70.4, 70.9, 71.1, 72.7, 72.9 (C2′′), 73.2, 74.5, 75.4, 76.0, 77.0, 78.1 (C2), 99.9 (C1′′′), 100.5 (C1′′), 100.8 (C1), 103.1 (C1′). HRMS (TOF): calc. for (M + Na)+ C27H49NO21Na 746.2695, found 746.2687.
Acknowledgements
Financial support from CSIR [02(0186)/14/EMR-II] to RG and from CAS-UGC and FIST-DST, India, to the Department of Chemistry, Jadavpur University are acknowledged. MMM (SRF) and NB (RA, CSIR project) thank UGC and CSIR, India, respectively, for their fellowships.References
- For review see B. L. Herwaldt, Lancet, 1999, 354, 1191 CrossRef CAS.
- http://www.who.int/topics/leishmaniasis/en.
- S. Sundar, S. G. Reed, V. P. Singh, P. C. Kumar and H. W. Murray, Lancet, 1998, 351, 563 CrossRef CAS.
- A. J. Magill, M. Grögl, R. A. Gasser, W. Sun and C. N. Oster, N. Engl. J. Med., 1993, 328, 1383 CrossRef CAS PubMed.
- J. Alvar, P. Aparicio, A. Aseffa, M. Den Boer, C. Cañavate, J.-P. Dedet, L. Gradoni, R. T. Horst, R. López-Vélez and J. Moreno, Clin. Microbiol. Rev., 2008, 21, 334 CrossRef CAS PubMed.
- S. M. Beverley and S. J. Turco, Trends Microbiol., 1998, 6, 35 CrossRef CAS PubMed.
- (a) A. Descoteaux, G. Matlashewski and S. J. Turco, J. Immunol., 1992, 149, 3008 CAS; (b) S. J. Turco and A. Descoteaux, Annu. Rev. Microbiol., 1992, 46, 65 CrossRef CAS PubMed.
- Y. Cabezas, L. Legentil, F. R. Gangneux, F. Daligault, S. Belaz, C. Nugier-Chauvin, S. Tranchimand, C. Tellier, J. P. Gangneux and V. Ferrières, Org. Biomol. Chem., 2015, 13, 8393 CAS.
- (a) S. J. Turco, S. R. Hull, P. A. Orlandi Jr, S. D. Shepherd, S. W. Homans, R. A. Dwek and T. W. Rademacher, Biochemistry, 1987, 26, 6233 CrossRef CAS PubMed; (b) S. J. Turco, P. A. Orlandi Jr, S. W. Homans, M. A. J. Ferguson, R. A. Dwek and T. W. Rademacher, J. Biol. Chem., 1989, 264, 6711 CAS; (c) P. A. Orlandi Jr and S. J. Turco, J. Biol. Chem., 1987, 262, 10384 CAS; (d) M. J. McConville, J. E. Thomas-Oates, M. A. J. Ferguson and S. W. Homans, J. Biol. Chem., 1990, 265, 19611 CAS.
- (a) S. W. Homans, A. Mehlert and S. J. Turco, Biochemistry, 1992, 31, 654 CrossRef CAS PubMed; (b) M. J. McConville, S. J. Turco, M. A. J. Ferguson and D. L. Sacks, EMBO J., 1992, 11, 3593 CAS.
- (a) B. J. Mengeling and S. J. Turco, Curr. Opin. Struct. Biol., 1998, 8, 572 CrossRef CAS PubMed; (b) B. J. Mengeling, S. M. Beverley and S. J. Turco, Glycobiology, 1997, 7, 873 CrossRef CAS PubMed; (c) M. J. McConville and M. A. J. Ferguson, Biochem. J., 1993, 294, 305 CrossRef CAS PubMed.
- (a) M. Kelleher, A. Bacic and E. Handman, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6 CrossRef CAS PubMed; (b) P. Talamas-Rohana, S. D. Wright, M. R. Lennartz and D. G. Russell, J. Immunol., 1990, 144, 4817 CAS; (c) D. M. Mosser, T. A. Springer and M. S. Diamond, J. Cell Biol., 1992, 116, 511 CrossRef CAS PubMed.
- A. Arasappan and B. Fraser-Reid, J. Org. Chem., 1996, 61, 2401 CrossRef CAS.
- M. Upreti, D. Ruhela and R. A. Vishwakarma, Tetrahedron, 2000, 56, 6577 CrossRef CAS.
- M. C. Hewitt and P. H. Seeberger, J. Org. Chem., 2001, 66, 4233 CrossRef CAS PubMed.
- M. C. Hewitt and P. H. Seeberger, Org. Lett., 2001, 3, 3699 CrossRef CAS PubMed.
- G. Sureshkumar and S. Hotha, Glycoconjugate J., 2012, 29, 221 CrossRef CAS PubMed.
- (a) S. K. Maity, S. Maity, A. Patra and R. Ghosh, Tetrahedron Lett., 2008, 49, 5847 CrossRef CAS; (b) S. K. Maity, A. Patra and R. Ghosh, Tetrahedron, 2010, 66, 2809 CrossRef CAS; (c) S. K. Maity, A. Patra and R. Ghosh, Synlett, 2012, 23, 1919 CrossRef CAS; (d) N. Basu, M. M. Mukherjee, A. Chaudhury and R. Ghosh, J. Indian Chem. Soc., 1815, 2013, 90 Search PubMed; (e) N. Basu, M. M. Mukherjee and R. Ghosh, RSC Adv., 2014, 4, 54084 RSC.
- G. Lemanski, T. Lindenberg, H. Fakhrnabavi and T. Ziegler, J. Carbohydr. Chem., 2000, 19, 727 CrossRef CAS.
- N. Basu, S. K. Maity and R. Ghosh, RSC Adv., 2012, 2, 12661 RSC.
- G. Zemplén, Ber. Dtsch. Chem. Ges., 1926, 59, 1254 CrossRef.
- (a) J. J. Gridley and H. M. I. Osborn, J. Chem. Soc., Perkin Trans. 1, 2000, 10, 1471 RSC; (b) A. Sau and A. K. Misra, PLoS One, 2012, 7, e37291 CAS.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS.
- (a) Y.-M. Zhang, A. Brodzky, P. Sinai, G. Saint-Marcoux and B. Perly, Tetrahedron: Asymmetry, 1995, 6, 1195 CrossRef CAS; (b) F. Yamazaki, S. Sato, T. Nukada, Y. Ito and T. Ogawa, Carbohydr. Res., 1990, 201, 31 CrossRef CAS PubMed.
- N. Basu, S. K. Maity, A. Chaudhury and R. Ghosh, Carbohydr. Res., 2013, 369, 10 CrossRef CAS PubMed.
- R. R. Schmidt, Angew. Chem., Int. Ed., 1986, 25, 212 CrossRef.
- (a) B. A. Garcia, J. L. Poole and D. Y. Gin, J. Am. Chem. Soc., 1997, 119, 7597 CrossRef CAS; (b) J. D. C. Codée, L. J. van den Bos, R. E. J. N. Litjens, H. S. Overkleeft, J. H. van Boom and G. A. van der Marel, Org. Lett., 2003, 5, 1947 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03856e |
| ‡ At present: Laboratory of Bioorganic Chemistry, Graduate School of Information Science, Nagoya University, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |