
Highly sensitive multifunctional recyclable Ag–TiO2 nanorod SERS substrates for photocatalytic degradation and detection of dye molecules†
 ‡a
‡a
Abstract
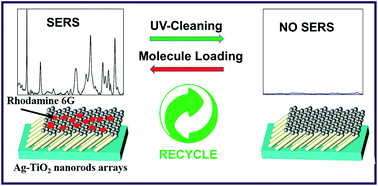
We report a facile method to fabricate novel Ag nanoparticle decorated TiO2 nanorod array substrates using a glancing angle deposition (GLAD) technique for photocatalysis and surface enhanced Raman scattering (SERS) applications. The silver nanoparticles on the TiO2 nanorods enhances the Raman signal of rhodamine 6G (Rh6G) dye with an enhancement factor ∼105. The photocatalytic degradation of Rh6G molecules present on Ag nanoparticle decorated TiO2 nanorods (Ag–TiO2) was also studied. The intensity of the Rh6G peak at 611 cm−1 decreases considerably (∼99%) after UV irradiation for 90 minutes. The change in SERS enhancement with time during ultra-violet (UV) light irradiation was used to track the photocatalytic degradation of the dye present on the Ag–TiO2 nanorods. The Ag–TiO2 SERS substrates can be recovered after 150 min in the presence of UV light illumination. The as-prepared Ag–TiO2 nanorods could be self-regenerated, fully recovered, and recycled after their use in the SERS analysis because of the self-cleaning property of TiO2 under simple UV light irradiation. This excellent SERS enhancement may be attributed to the formation of high density hotspots on top of the TiO2 nanorods which is supported by finite difference time domain (FDTD) simulations.
 Please wait while we load your content...
Please wait while we load your content...

