
A theoretical investigation of the removal of methylated arsenic pollutants with silicon doped graphene†
Diego Cortés-Arriagada* and
Alejandro Toro-Labbé
Nucleus Millennium Chemical Processes and Catalysis, Laboratorio de Química Teórica Computacional (QTC), Departamento de Química-Física, Facultad de Química, Pontificia Universidad Católica de Chile, Av. Vicuña Mackenna 4860, Macul, Santiago 9900087, Chile. E-mail: dcortesr@uc.cl
First published on 11th March 2016
Abstract
Quantum chemistry calculations were performed to study the adsorption of methylated arsenic pollutants onto silicon doped graphene (SiG), characterizing their geometrical parameters, energetic and binding properties. All the methylarsenicals are chemisorbed onto SiG by silicon–oxygen binding. Trivalent methylarsenicals are chemisorbed with adsorption energies of up to 1.1 eV at neutral conditions; while, pentavalent methylarsenicals reach energies of up to 1.8 and 4.0 eV in their neutral and anionic states, respectively. Additionally, it was determined that the pollutant stability after the chemisorption plays a key role in the adsorption strength; contributions from long-range interactions and charge transfer processes were also discussed. Finally, molecular dynamics and explicit solvent simulations indicated that the adsorption stability remains stronger both in aqueous environments and at ambient conditions (300 K); in water environments at neutral pH, trivalent and pentavalent methylarsenicals will be adsorbed mainly in their neutral and anionic forms, respectively. Therefore, SiG emerges as a promising material for technologies related to the removal of organic arsenic pollutants in their trivalent and pentavalent oxidation states.
1. Introduction
Nowadays, arsenic compounds correspond to a priority class of pollutants whose presence in natural sources constitutes a critical environmental problem; the WHO (World Health Organization) established the standards for arsenic in drinking water and air quality as 10 μg L−1 and 1.5 × 10−3 per μg m−3, respectively.1,2 Nevertheless, these standards are exceeded in many places of the planet due to the anthropogenic and industrial activities, or by the geochemical nature of the ground. The methylated forms of arsenic appear as part of the arsenic cycle in the environment, or from the arsenic metabolism in humans and animals.3 The exposure to methylated arsenic containing waters has been associated with alterations in the birth weight and gestational age,4 skin lesions,5 lung and bladder cancer,6 renal dysfunctions,7 among other effects on the human health. Due to the high cellular uptake of the trivalent methylarsenicals, their toxicity is even larger than those determined for inorganic trivalent arsenic (iAsIII).8,9 Moreover, although the cellular uptake is lower for the pentavalent methylarsenicals with respect to inorganic pentavalent arsenic (iAsV), their toxicity is dangerous in long periods, and they are also reported as carcinogens.10,11 Therefore, the development of new materials for the control and removal of methylarsenicals from the environment becomes an important effort.In recent years, graphene has emerged as a useful material for the development of new adsorbent materials for extraction of several organic and inorganic pollutants;12,13 due to that its lamellar structure, graphene allows a large surface area available for pollutant adsorption, improving the performance, for instance, in wastewater treatment.12–14 For the arsenic removal, intrinsic graphene has been determined with low efficiency from water sources because of the weak adsorbent–adsorbate interactions;13,15 however, modified graphene (and oxidized graphene) composites have been reported as efficient adsorbent materials for the removal and/or detection of arsenic compounds, with high adsorption capacities from drinking waters and low adsorbent aggregation.16–21 In this regard, density functional theory (DFT) studies have shown the ability of silicon embedded graphene (SiG) for the adsorption of a series of molecules such as hydrogen cyanide,22 nitrogen oxides,23,24 formic acid,25 oxygen,24,26 water,24 and carbon monoxide;24,27 besides, the silicon doping increases the hydrogen storage in graphene.28 Furthermore, we have recently shown from DFT calculations that SiG also shows good adsorbent properties for the removal of inorganic trivalent arsenic even in water environments,15 with adsorption energies of ∼1.0 eV.
Although only a few experimental efforts were developed to obtain silicon doped graphene, some recent works have reported its successful obtaining. Lv and co-workers achieved the chemical synthesis of large-area SiG sheets from methoxytrimethylsilane (silicon precursor) and hexane (carbon precursor), which were grown over copper substrates; the SiG sheets showed an enhanced performance for the adsorption and sensing and the Raman scattering of organic dyes.29 In addition, Wang and co-workers obtained SiG by means of the chemical vapour deposition technique, using triphenylsilane as a carbon and silicon precursor; SiG was also grown onto a copper substrate.30 In this sense, new technological applications of SiG are expected to be announced in the coming years.
Taking into account the rise of SiG and its potential applications, and the development of new adsorbent materials for arsenic removal, we study by means of DFT calculations the removal of methylated arsenic pollutants using SiG sheets. The adsorption of trivalent and pentavalent forms of methylated arsenicals onto SiG was fully characterized from its geometrical parameters, energetic and binding properties. Solvent effects and molecular dynamics studies were also performed to account for the removal efficiency at water and ambient conditions.
2. Computational methodology
A C94H24 finite lattice was adopted as a graphene surface model, where the dangling bonds at the edges were saturated with hydrogen atoms; the silicon doped graphene (SiG) was built by replacing one carbon atom by one Si atom (SiC93H24). The adsorption energies were well converged onto this adsorbent model, so that it was selected for the calculations. The dopant concentration was retained below 5%, because only a silicon doping content of ∼1.75–2.63% has been experimentally reached.29,30 As trivalent methylarsenicals, we study the water soluble trivalent species monomethylarsonous acid [MMAIII, CH3As(OH)2] and dimethylarsinous acid [DMAIII, (CH3)2AsOH], which are more toxic than the inorganic form, this is arsenous acid [iAsIII, As(OH)3]. As pentavalent methylarsenicals, we use monomethylarsonic acid [MMAV, CH3AsO(OH)2] and dimethylarsinic acid [DMAV, (CH3)2AsO(OH)], and comparisons were performed with inorganic pentavalent arsenic or arsenic acid [iAsV, AsO(OH)3]. Neutral and anionic forms of pentavalent arsenicals were studied, where the anionic forms of iAsV, MMAV and DMAV are called arsenate, methylarsenate and dimethylarsenate, respectively.All the DFT calculations were performed in the ORCA 3.0 program31 with the Perdew–Burke–Ernzerhof (PBE) functional;32 the usage of the PBE method has been reported to be adequate in the study of the arsenic adsorption onto mineral substrates such as iron and titanium oxides,33,34 and even onto intrinsic and doped graphene in good agreement with more expensive ab initio methodologies.15 The all-electron Ahlrichs polarized double zeta basis sets (SVP) were used for H, C, O and Si atoms; a polarized triple-zeta basis set (TZVP) was adopted for As. The pairwise dispersion correction DFT-D3 was used to include dispersion force effects on energy and gradients, in combination with the Becke–Johnson damping scheme in order to avoid repulsive interatomic forces at short distances.35,36 Geometry optimizations and SCF steps were converged with tolerance values of 1 × 10−6 and 1 × 10−8 Hartree, respectively. The COSMO solvent model was implemented in selected calculations,37 with water as solvent; the COSMO calculations were carried-out in the ORCA 3.0 program.
Atomic net charges were obtained from the natural population analysis performed in the NBO 6.0 program;38 in these cases, single point calculations were performed in the ORCA 3.0 program with the NPA keyword in order to obtain the .47 file for all the systems; the .47 file contains information about the basis set, the density matrix, the Fock matrix, among others, which are used by the NBO 6.0 program to obtain properties as natural charges and natural bond orbitals. Wavefunction and Atoms-in-Molecules (AIM) analyses were carried-out in the Multiwfn analyzer;39 in these cases, the .gbw files (which are generated from the ORCA calculations) were converted to .molden files by means of the orca_2mkl program included in the ORCA 3.0 package; then, the .molden files were used as input files for the Multiwfn code.
For all the adsorbent–adsorbate systems, the adsorption energies (Eads) were obtained as:
| Eads = Eadsorbent + EP − Eadsorbent+P + EvdW | (1) |
The adsorbate–adsorbent stability was analyzed by means of ab initio molecular dynamic (MD) calculations with the ADMP method (atom density matrix propagation),41–43 which allows to obtain the propagation of both the nuclear centers and electron density; thus, giving an adequate behavior of the chemical bonding in dynamic conditions. The potential was determined “on-the-fly” at the PBE/3-21G* level of theory (a minimal basis set was implemented due to the high computational cost). The time step for all the simulations was of 0.1 fs, and 1.0 ps was used for statistics; control of temperature was reached by velocity scaling. ADMP-MD calculations were performed in the Gaussian09 program44 from optimized molecular structures in the ORCA 3.0 program.
3. Results and discussion
3.1 Adsorption of MMAIII and DMAIII
In the first place, the trivalent arsenicals are deprotonated above pH = 9.2; then, trivalent arsenicals are neutrally charged at neutral and physiological pH.3,35,36 For that reason, the neutral forms of MMAIII and DMAIII were selected for the calculations (Fig. 1a).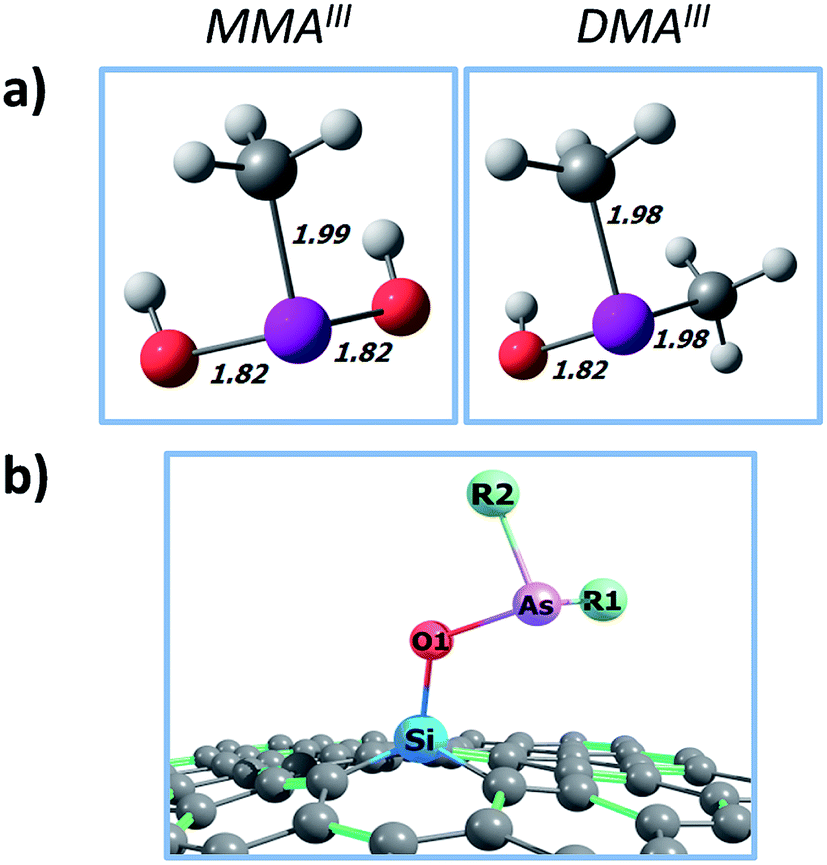 | ||
| Fig. 1 (a) Optimized molecular structures of MMAIII and DMAIII; distances in angstroms (Å); color code by atom: white (H); grey (C); red (O); purple (As). (b) General sketch for the interaction of trivalent methylarsenicals onto SiG; R1 and R2 groups change between MMAIII and DMAIII; green lines stand for double bonds, and hydrogen atoms were omitted in the molecular representation. | ||
In Fig. 1 are depicted the optimized molecular structures of the SiG–MMAIII and SiG–DMAIII systems, where three (a–c) and two (a and b) stable conformations were obtained, respectively. Two geometrical orientations were found; (i) where the pollutant acquires a “lying-down” orientation onto SiG (conformation a), with all the As–OH and As–CH3 bonds directed to the substrate plane; and (ii) where the pollutant acquires a “seated” orientation onto SiG (conformations b and c), with one As–OH (or As–CH3) bond directed away from the adsorbent plane. In all the cases, the adsorbent–adsorbate interactions take place through a Si–O bond (chemisorption), with the bond lengths in the range of dSi–O1 = 1.83–1.88 Å (Fig. 2). No interactions were obtained with the arsenic moiety, with Si⋯As distances over 3.3 Å, which agree with the substrate–As distances in the arsenic adsorption onto mineral substrates (3.2–3.5 Å) as determined from EXAFS experiments.45,46 In addition, in the free MMAIII and DMAIII, the As–OH and As–CH3 bonds show distances of ∼1.8 and ∼2.0 Å, respectively; although the As–CH3 bonds remain almost unchanged after the chemisorption, the interacting As–OH bond is elongated in up to ∼0.3 Å, thus, slightly destabilizing the pollutant structure.
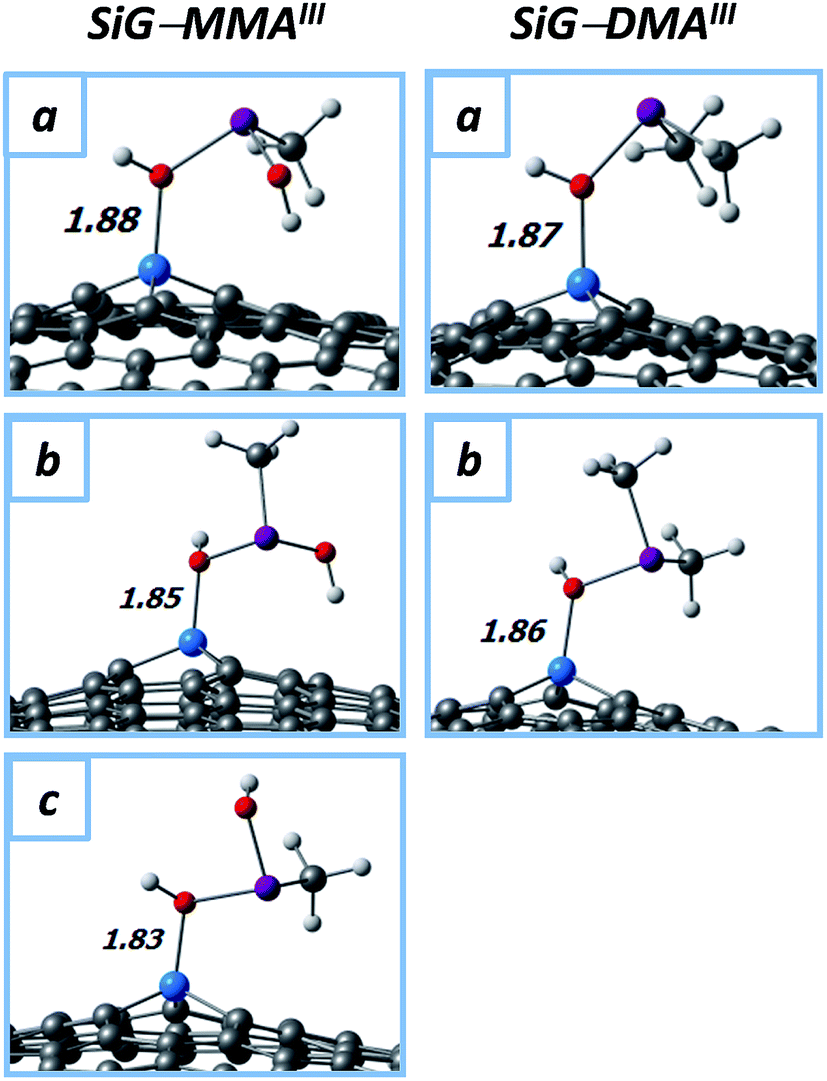 | ||
| Fig. 2 Molecular structures of MMAIII and DMAIII adsorbed onto SiG; up to three conformations were obtained per system (a–c). Distances are in angstroms (Å). Color code: white (H); grey (C); red (O); sky-blue (Si); purple (As). | ||
With regard to the adsorption strength, in Table 1 are listed the adsorption energies (Eads) of the different SiG–pollutant conformations. As observed in Table 1, MMAIII and DMAIII show similar Eads values, which are ranging from 0.9 to 1.1 eV; there are not significant differences between the adsorption strength of the different conformations, with differences of up to 0.15 eV. The adsorption strength is similar to that of inorganic trivalent arsenic (iAsIII) onto SiG (∼1.0 eV),15 indicating that the adsorption of either iAsIII, MMAIII or DMAIII will be stable onto SiG. It is worth noting that in addition to the chemical binding, the dispersion forces contribute to the stability of all these systems (EvdW and QP→G parameters in Table 1). The contribution of dispersion forces is ∼0.4 eV, which represents up to 42% of the adsorption energy (∼0.4 eV). It was also observed that the chemisorption process is accompanied by an electron donation of ∼0.2|e| from the methylarsenicals towards the SiG sheet.
Another relevant point is that the reliability of the PBE adsorption energies was evaluated against those obtained with the hybrid B3LYP-D3 method in combination with the same basis sets as implemented for PBE. These results are displayed in Table 1 (in parenthesis), and it was found that the differences between both methods are less than 0.10 eV, indicating the accuracy of the selected methodology without a significant increase in the computational cost.
At this point, it is important to highlight that the adsorption of trivalent arsenicals onto the SiG graphene shows a reasonable performance in comparison with mineral substrates. For instance, Oliveira and co-workers47 determined the arsenite adsorption onto aluminum oxides (gibbsite) with adsorption energies of ∼0.64 eV from DFT calculations; Blanchard and co-workers33 showed from DFT calculations that the bidentate interaction of arsenite with FeS2 takes place with an adsorption energy of ∼1.44 eV; moreover, Wei and co-workers48 has determined the arsenite adsorption onto TiO2 anatase (which is a well know to effectively remove arsenic from polluted waters) with adsorption energies of up to ∼0.80 eV at neutral conditions. In summary, SiG show good adsorbent properties for the removal of organic and inorganic trivalent methylarsenicals. On the other hand, the industrial activity is the main source of atmospheric arsenic and responsible for the presence of methylarsenicals in the particulate matter,49–51 so that the proposed adsorbents could be also applied for removal of methylarsenicals in the gas phase or its detection.
3.2 Adsorption of MMAV and DMAV
Additionally, we also studied the ability of SiG to adsorb pentavalent methylarsenicals (MMAV, DMAV). Also, the adsorption of inorganic pentavalent arsenic (iAsV) was studied for comparison purposes and completeness of the work. It is worth to note that the pentavalent arsenicals have different acid–base equilibrium depending on the pH. For instance, iAsV is a triprotic acid and dissociates in water according to:3,52| AsO(OH)3 ⇔ AsO2(OH)2− + H+, pKa1 = 2.3 | (2) |
| AsO2(OH)2− ⇔ AsO3(OH)2− + H+, pKa1 = 6.8 | (3) |
| AsO3(OH)2− ⇔ AsO43− + H+, pKa1 = 11.6 | (4) |
Therefore, almost equal forms of arsenate AsO2(OH)2− and AsO3(OH)2− could exist at neutral pH. Likewise, MMAV and DMAV behave as diprotic and monoprotic acids, respectively, with the equilibrium:3,53
| CH3AsO(OH)2 ⇔ CH3AsO2(OH)− + H+, pKa1 = 4.1 | (5) |
| CH3AsO2(OH)− ⇔ CH3AsO32− + H+, pKa1 = 8.7 | (6) |
| (CH3)2AsO(OH) ⇔ (CH3)2AsO2− + H+, pKa1 = 6.2 | (7) |
Consequently, the anionic form of MMAV [CH3AsO2(OH)−] is the main one at neutral pH; conversely, both neutral and anionic species of DMAV (CH3)2AsO(OH) and (CH3)2AsO2− are capable to exist at neutral pH. Taking into account these considerations, we performed calculations onto the anionic forms, which are expected to be the main species adsorbed onto SiG at neutral conditions in aqueous environments (Fig. 3a). The neutral forms were also computed because they are important to get insights about the removal ability in a broad range of pH, as well as for the arsenic adsorption in the gas phase.
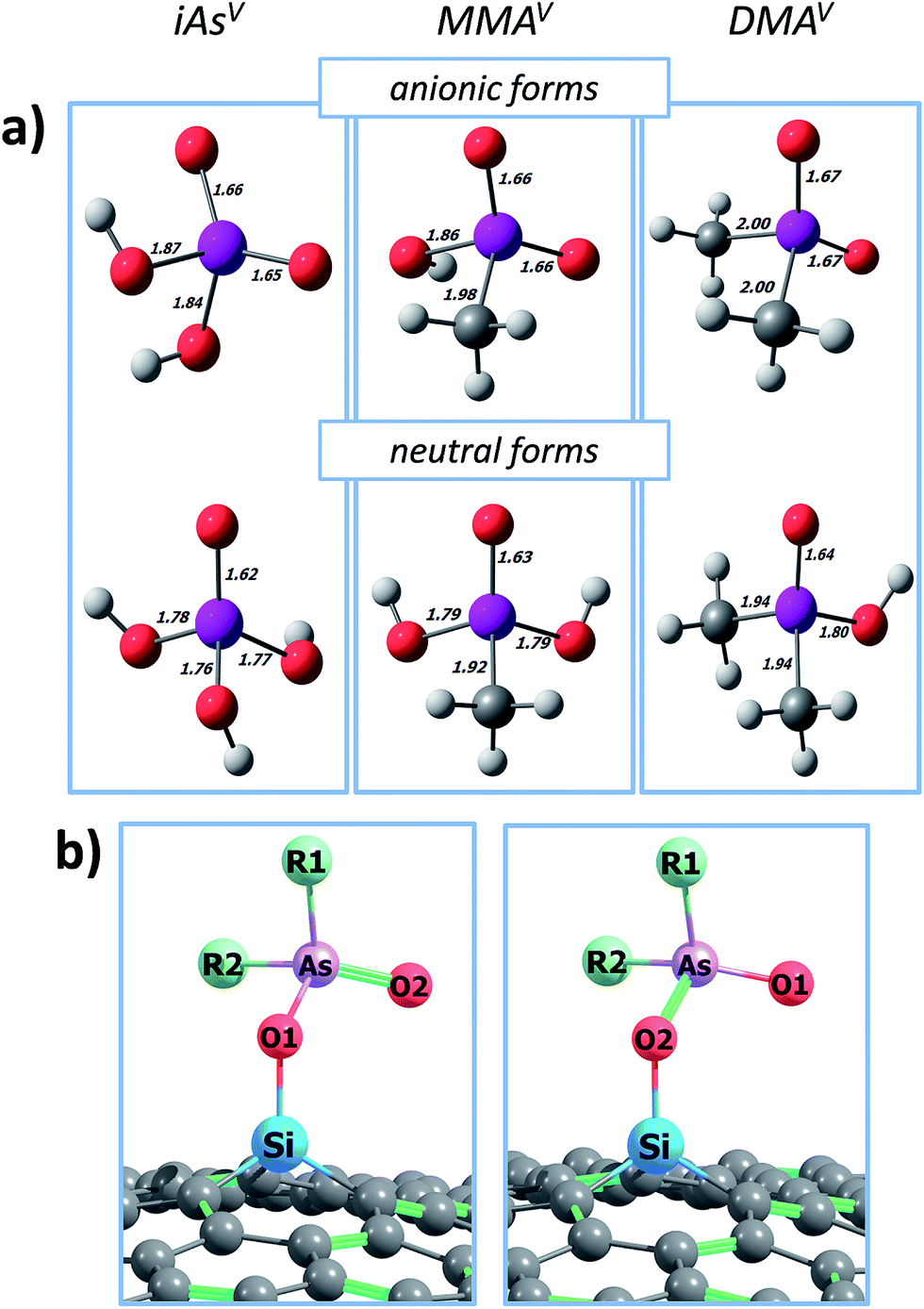 | ||
| Fig. 3 (a) Optimized molecular structures of iAsV, MMAV and DMAV at their anionic and neutral forms; distances in angstroms (Å); color code by atom: white (H); grey (C); red (O); purple (As). (b) General sketch for the interaction of pentavalent arsenicals onto SiG; R1 and R2 groups change between iAsV, MMAIII and DMAIII; green lines stand for double bonds, and hydrogen atom were deleted to simplify. | ||
In the first place, the optimized molecular structures of the anionic MMAV and DMAV adsorbed onto SiG are displayed in Fig. 4, in addition to those for iAsV for comparison. Two (a and b), three (a–c) and two (a and b) conformations were found for the adsorption of iAsV, MMAV and DMAV, respectively, where a “seated” orientation is adopted. In all the cases, silicon binds to one deprotonated oxygen atom of the pollutant (which is labeled as O1 in Fig. 3b), with the bond lengths of dSi–O ≈ 1.7 Å; note that these distances are at least ∼0.1 Å shorter than in the adsorption of the trivalent forms. The latter suggest that the adsorption strength could be sorted as DMAV > MMAV > iAsV, and even that the adsorption energies would be higher than for trivalent methylarsenicals. Indeed, from Table 2, it is noted an important increasing in the adsorption energies, with values of up to 4.01 eV. Specifically, the chemisorption of iAsV, MMAV and DMAV is characterized with Eads values on the range of 3.31–3.34 eV, 3.63–3.64 eV and 3.93–4.01 eV, respectively, which agree with the slight differences in the Si–O1 bond lengths, and sorting the chemisorption strength as DMAV > MMAV > iAsV (in other words, the adsorption strength increases as the methyl groups in the pollutant increases). Note that the interacting As–O1 bond distances in the anionic MMAV and DMAV are slightly affected due to the formation of the Si–O1 interaction, with the bond lengths of dAs–O1 ≈ 1.80 Å; the latter indicates that the σAs–O1 bond is not weakened due to the chemisorption as observed in the case of MMAIII/DMAIII, and retaining the pollutant stability.
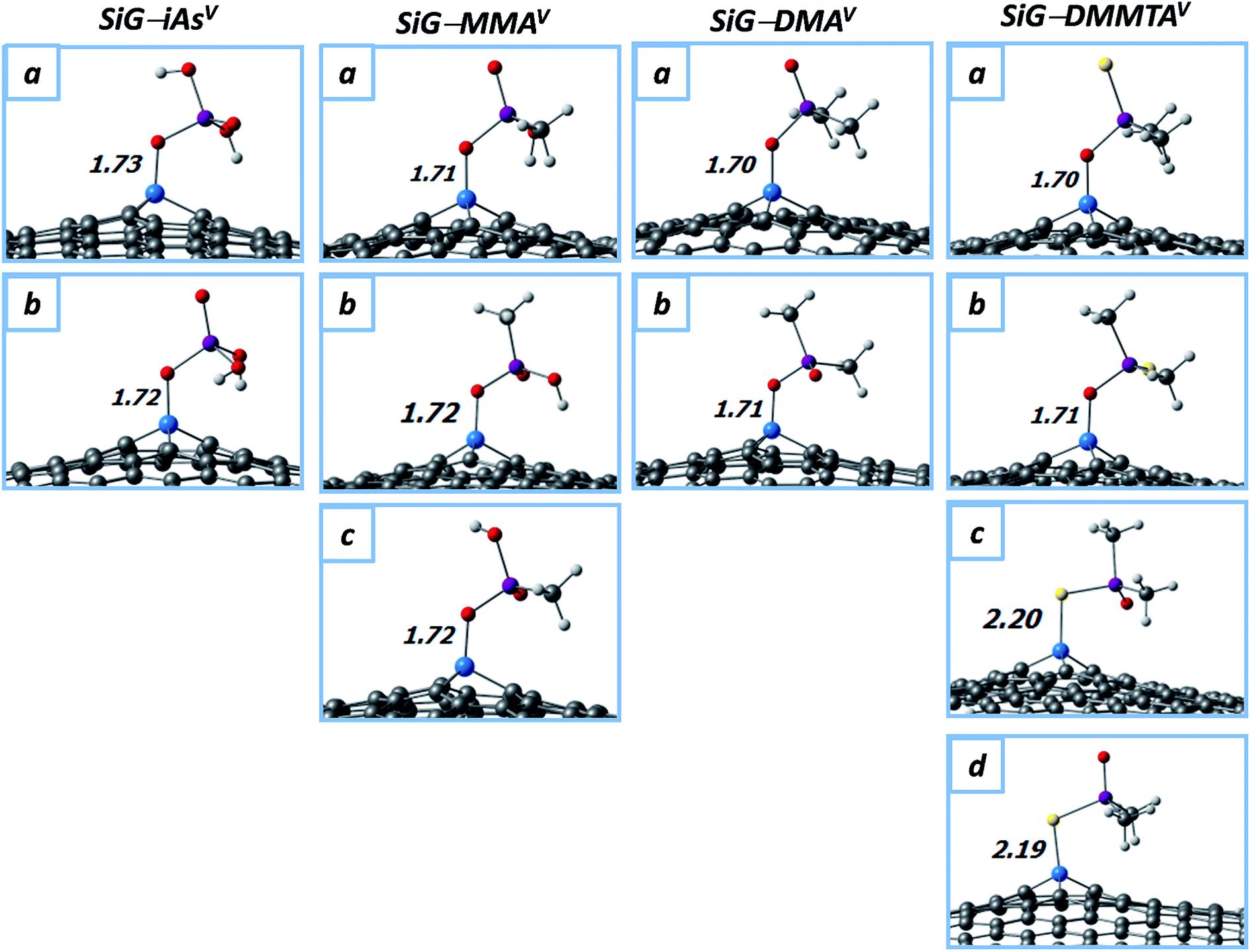 | ||
| Fig. 4 Molecular structures of iAsV, MMAV, DMAV and DMMTAV (in their anionic forms) adsorbed onto SiG; up to four conformations were obtained per system (a–d). Distances are in angstroms (Å). Color code: white (H); grey (C); red (O); sky-blue (Si); yellow (S); purple (As). | ||
| Pollutant | Eads | EvdW | QP→G | dSi–O1 | dAs–O1 | |
|---|---|---|---|---|---|---|
a Adsorption energies computed with the minimally augmented def2 basis sets (ma-def2-SVP).b For DMMTAV, these interaction modes correspond to Si–S binding.c In these conformations of DMMTAV, this bond corresponds to As–S.d In these cases, the interaction with silicon is with the As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2 moiety as showed in Fig. 3b. O2 moiety as showed in Fig. 3b. |
||||||
| Anionic pentavalent arsenicals | ||||||
| iAsV | a | 3.31 | 0.35 | 0.40 | 1.73 | 1.78 |
| b | 3.33 | 0.37 | 0.41 | 1.72 | 1.77 | |
| MMAV | a | 3.64 (3.76)a | 0.40 | 0.42 | 1.71 | 1.79 |
| b | 3.64 (3.70)a | 0.36 | 0.41 | 1.72 | 1.78 | |
| c | 3.63 (3.67)a | 0.36 | 0.42 | 1.72 | 1.78 | |
| DMAV | a | 4.01 (4.14)a | 0.42 | 0.43 | 1.70 | 1.79 |
| b | 3.93 (4.04)a | 0.38 | 0.42 | 1.71 | 1.80 | |
| DMMTAV | a | 3.79 | 0.43 | 0.42 | 1.70 | 1.80 |
| b | 3.72 | 0.43 | 0.41 | 1.71 | 1.80 | |
| c | 2.96 | 0.47 | 0.68 | 2.20b | 2.28c | |
| d | 3.08 | 0.49 | 0.72 | 2.19b | 2.28c | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Neutral pentavalent arsenicals | ||||||
| iAsV | a | 0.67 | 0.44 | 0.16 | 1.89 | 1.95 |
| b | 1.52 | 0.45 | 0.19 | 1.73d | 1.82d | |
| MMAV | a | 0.71 | 0.44 | 0.20 | 1.89 | 1.97 |
| b | 0.58 | 0.40 | 0.19 | 1.94 | 1.95 | |
| c | 0.57 | 0.43 | 0.16 | 1.89 | 1.96 | |
| d | 1.58 | 0.47 | 0.23 | 1.79d | 1.73d | |
| e | 1.66 | 0.44 | 0.24 | 1.79d | 1.73d | |
| DMAV | a | 1.23 | 0.45 | 0.20 | 1.88 | 1.97 |
| b | 1.73 | 0.48 | 0.23 | 1.78d | 1.74d | |
| c | 1.77 | 0.45 | 0.26 | 1.79d | 1.73d | |
With respect to the contribution of dispersion forces (EvdW) to the adsorption strength, it can be observed in Table 2 that they are not so important, with contributions of up to 0.49 eV (∼10% of the total Eads). On the other hand, it is clear that the anionic nature of MMAV and DMAV increases the amount of electron transfer towards the acceptor SiG in up to 100% with respect to the trivalent forms. The electron transfer is similar for each the arsenic compounds, where ∼0.4|e| are transferred to the adsorbent.
It must be also noted that due to the anionic nature of the pollutants, the accuracy of the adsorption energies was evaluated against those obtained by using a diffuse basis set. The minimally augmented Karlsruhe def2 basis sets (ma-def2-SVP)54 were used, which include diffuse s and p functions for all the atoms; these calculations were performed only with anionic MMAV and DMAV due to the high computational cost. Note that other diffuse basis sets showed problems with linear dependences and convergence. From Table 2 (in parenthesis), we observe that only differences of up to 0.13 eV are obtained in the Eads values comparing both basis sets, indicating the good accuracy of the Eads values.
Dimethylmonothioarsinic acid (DMMTAV) is another common methylated pollutant, whose presence in the environment is favored by the metabolic process of mammals, in addition to the sulfidic nature of grounds.55–58 The cytotoxicity of DMMTAV is larger than DMAV, and similar to DMAIII, and its toxicity has been associated with the production of reactive oxygen molecules during its exposure, which could cause DNA damage in order to initialize cancer.3,10 Taken into account these considerations, we carried-out calculations with anionic DMMTAV (dimethylmonothioarsenate) as adsorbate to get insights into the removal of methylated thioarsenicals. The SiG–DMMTAV conformations are also displayed in Fig. 4, and their properties are quoted in Table 2. Four interaction modes were found (a–d), where the chemisorption takes place by means of Si–O (a and b) or Si–S (c and d) binding, with bond distances of dSi–O ≈ 1.7 Å and dSi–S ≈ 2.2 Å, respectively. The chemisorption is favored in the Si–O mode with Eads values of up to 3.79 eV; while, the Si–S conformations reach Eads value of up to 3.08 eV. Consequently, the efficiency of SiG would be good for a broad kind of trivalent and pentavalent arsenicals, also including their thiolated forms.
We studied the adsorption of neutral iAsV, MMAV and DMAV to get insights about the removal at low pH or gas phase (see Fig. 5 and Table 2). In these cases, the chemisorption takes place by two binding modes. In the first binding mode (conformations a, a–c, and a for iAsV, MMAV and DMAV, respectively), the silicon atom interacts with the protonated oxygen belonging to one σAs–O1 bond of the pollutant, with bond distances of dSi–O1 ≈ 1.90 Å. The adsorption energies reach values of up to 0.67, 0.71 and 1.23 for iAsV, MMAV and DMAV, respectively, which are similar in comparison with adsorption of trivalent methylarsenicals. Note that in these conformations, the As–O1 bond it is elongated from −1.8 to −2.0 Å, suggesting the depletion of the σAs–O1 bond as for trivalent arsenicals.
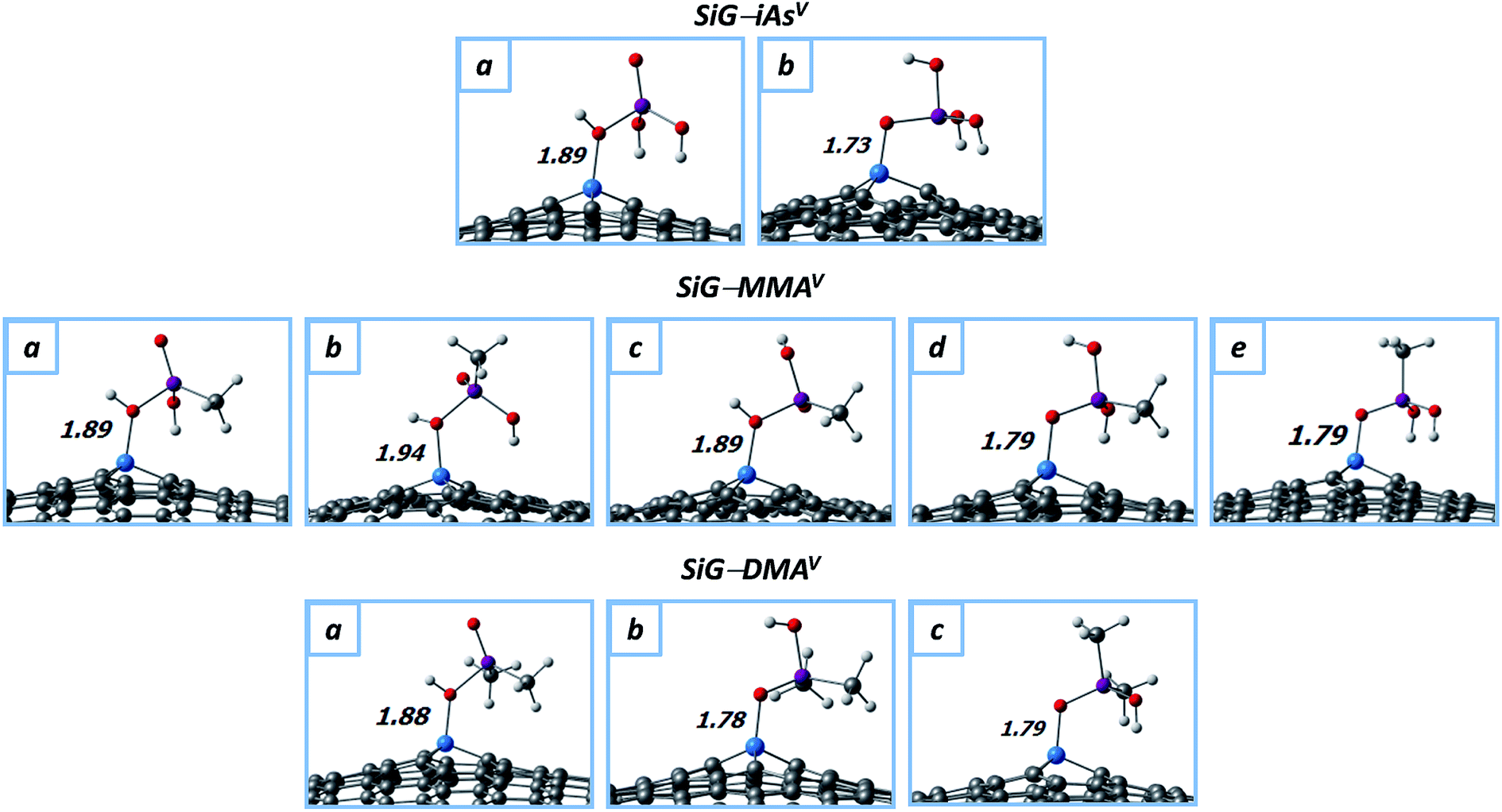 | ||
| Fig. 5 Molecular structures of iAsV, MMAV and DMAV (in their neutral forms) adsorbed onto SiG; up to five conformations were obtained per system (a–e). Distances are in angstroms (Å). Color code: white (H); grey (C); red (O); sky-blue (Si); purple (As). | ||
Conversely, in the second binding mode, silicon binds to the deprotonated oxygen atom belonging to the πAsO2 bond of the pollutant (conformations c–e, and b and c for iAsV, MMAV and DMAV, respectively). These conformations are favored with short bond lengths in the range of dSi–O1 = 1.73–1.79 Å, in addition to enhanced adsorption energies of up to 1.52, 1.66 and 1.77 eV for iAsV, MMAV and DMAV, respectively. In these cases, the double πAsO2 bond is elongated from ∼1.63 to ∼1.73 Å for MMAV and DMAV, which is near to distances for a single As–O bond. Then, the πAsO2 bond would be weakened to allow the Si–O2 interaction, but without a major structural destabilization of the pollutant because of the As–O2 bond remains still stable by single bonding. Likewise, in the case of iAsV, the presence of three electron-withdrawing –OH groups in the pollutant structure cause a slight larger depletion of the πAsO2 bond density with respect to the methylated arsenicals, increasing its bond distance from 1.62 to 1.82 Å. Finally, these results indicate that the adsorption strength of neutral arsenicals onto SiG can be sorted as DMAV > MMAV > iAsV > DMAIII ≈ MMAIII ≈ iAsIII; this finding might to serve as a guidance for experimental studies.
In summary, the adsorption of pentavalent methylarsenicals (inorganic and organic) appears highly improved onto SiG with respect to the trivalent forms, so that a promising new material for arsenic removal is proposed with efficiency from neutral to low pH conditions. The latter is also in agreement with the lower mobility of pentavalent arsenicals in comparison with those obtained for the trivalent species when they are adsorbed from aqueous solutions, which also causes the high toxicity of the trivalent forms.59,60 As noted above, the anionic and neutral pentavalent methylarsenicals (also including arsenate) are adsorbed with energies of up to 4.01 and 1.77 eV, respectively, which constitutes an important improvement in comparison to other adsorbents. For instance, the arsenate adsorption onto TiO2 based surfaces (such as anatase and rutile) has been determined with adsorption energies of up to ∼3.0 eV;34,61,62 Ladeira and co-workers estimated in ∼2.6 eV the arsenate chemisorption onto gibbsite (an aluminum hydroxide);45 the arsenate adsorption onto mineral iron oxides (such as goethite, lepidocrocite and hematite) reached a value of ∼2.4 eV;63 while, iAsV and DMAV are chemisorbed onto Fe-(oxyhydr)oxides with adsorption energies of up to ∼1.6 eV.64
With regard to the recovery of SiG after the removal, it is known that the adsorption of anionic pollutants is competitive in alkaline conditions with the adsorption of hydroxyl anions; the latter avoids the uptake of anionic pollutants because the hydroxyl groups turn negatively charged the adsorbent surface, and causing a repulsive coulombic interaction with the anionic arsenicals.13 Then, the recovery of the adsorbent material would be straightforwardly reached by means of high pH solutions. On the other hand, a high recovery of doped graphene adsorbents has been reached by thermal treatment; for instance, thermal treatment up to 800 °C allows the desorption of adsorbed thiophene (84%) from P-doped graphene, without thermal degradation of the substrate.65
3.3 Atoms in molecules (AIM) analysis
In this section, we present some interesting features determining the strength of the SiG–pollutant interactions. We used the electron density (ρi) at the bond critical points (BCPs) as obtained from the AIM methodology, which are listed in Table 3. To take into consideration, note that due to the different electronegativity values of C, O and As atoms [2.55 (C), 3.44 (O) and 2.18 (As) in the Pauling scale], the electron bond densities at the BCP of the As–C and As–O bonds (ρSi–O1 and ρAs–C) are lower than 0.2 a.u. because the bonds have a relative high ionic character;66 in addition, the Si–O bond corresponds to a closed-shell interaction,66 so that the electron density at the BCP will be of ∼0.1 a.u.| Pollutant | ρSi–O1 | ρAs–O1 | ρC⋯H | ρC⋯H′ | |
|---|---|---|---|---|---|
| Neutral trivalent arsenicals | |||||
| MMAIII | a | 0.074 | 0.086 | 0.010 | 0.023 |
| b | 0.079 | 0.082 | 0.023 | — | |
| c | 0.082 | 0.077 | 0.013 | 0.020 | |
| DMAIII | a | 0.074 | 0.091 | 0.008 | 0.013 |
| b | 0.077 | 0.087 | 0.012 | — | |
|
|||||
| Anionic pentavalent arsenicals | |||||
| MMAV | a | 0.107 | 0.145 | 0.020 | 0.011 |
| b | 0.106 | 0.148 | 0.013 | — | |
| c | 0.105 | 0.149 | 0.005 | 0.007 | |
| DMAV | a | 0.109 | 0.142 | 0.008 | 0.011 |
| b | 0.107 | 0.143 | 0.007 | 0.005 | |
|
|||||
| Neutral pentavalent arsenicals | |||||
| MMAV | a | 0.071 | 0.098 | 0.028 | 0.008 |
| b | 0.066 | 0.104 | 0.021 | — | |
| c | 0.070 | 0.102 | 0.008 | — | |
| d | 0.089 | 0.169 | 0.029 | 0.008 | |
| e | 0.088 | 0.171 | 0.026 | 0.019 | |
| DMAV | a | 0.072 | 0.097 | 0.015 | 0.006 |
| b | 0.091 | 0.164 | 0.008 | 0.008 | |
| c | 0.088 | 0.169 | 0.023 | 0.013 | |
First, when MMAIII and DMAIII are adsorbed onto SiG, the interacting As–O1H bond is elongated up to ∼0.3 Å. In this regard, the electron density at the BCP of the As–O1 bond (ρAs–O1) shows values of 0.08–0.09 a.u., indicating that the single bond is weakened in up to ∼40% due to the chemisorption with respect to the free pollutants (ρAs–O1 ≈ 0.14 a.u.). In addition, the electron density at the BCP of the Si–O1 bond are on the range of ρSi–O1 = 0.07–0.08 a.u., which are lower than for a single covalent bond due to the coordinative covalent character of this interaction. These results clearly suggest that the in the SiG–pollutant interaction, the pollutant mainly transfers electron density from the interacting As–O1 bond toward the new formed Si–O1 bond, causing the destabilization of the pollutant structure and limiting the adsorption strength.
With regard to the free MMAV and DMAV, the deprotonation of one –OH group causes the improving and weakening of the σAs–O1 and πAsO2 bonds, respectively, due to resonance effects. For instance, from Fig. 3a, it is observed that for the neutral DMAV molecule, the As–O1 and As–O2 bond distances are about 1.80 and 1.64 Å, respectively, showing a double and single bond, respectively; whereas, in the free anionic DMAV, the As–O1 and As–O2 bonds are equivalents with distances of 1.67 Å and bond order of ∼1.73, so that the electron density excess is delocalized trough the σAs–O1 and πAsO2 bonds to form two resonant double bonds.
The above has an impact on the adsorption strength of anionic pentavalent arsenicals onto SiG (and probably onto other substrates). In these cases, the ρAs–O1 values after the chemisorption remain in ∼0.14 a.u., indicating that the bond turns more covalent than ionic and retains the single bond nature unlike in the adsorption of trivalent arsenicals. The negative charge excesses also increase the stability of the Si–O1 bond, increasing its density up to ρSi–O1 ≈ 0.11 a.u. Therefore, the high adsorption energies obtained for the chemisorption of anionic MMAV and DMAV are explained because the interacting As–O1 bond of the pollutant is strengthened before the interaction due to resonant effect, showing a quasi double bonding; then, when chemisorption is reached, the quasi double As–O1 bond transfer bond density to strengthen the Si–O1 bonding, but the final As–O1 bond is capable to retain a single nature.
Finally, in the case of neutral pentavalent arsenicals, the most stable conformations take place by binding of silicon with the deprotonated oxygen atom belonging to the pollutant πAsO2 bond (conformations d and e for MMAV, and b and c for DMAV). In these cases, the electron density at the BCP indicates that the double πAsO2 bond is weakened to allow the Si–O2 interaction without a major structural destabilization of the pollutant because of the As–O2 bond remains still stable by single bonding, thus causing a higher adsorption energy with respect to the interaction by the σAs–O1 bond (in these cases the behavior is similar to trivalent methylarsenicals). For instance, in conformation “c” of the adsorption of neutral DMAV onto SiG, the interacting πAsO2 bond is weakened from the double bond in the free pollutant (ρAs–O1 = 0.21 a.u.) until an almost single σAs–O2 bond (ρAs–O1 = 0.17 a.u.) after chemisorption, which helps to retain the pollutant stability, in addition to slightly increase the strength of the Si–O bond (ρAs–O1 = 0.09 a.u.) as in the case of the anionic pentavalent methylarsenicals.
In summary, after the SiG–pollutant interactions, the stability of the σAs–O or πAsO bonds of the methylarsenicals plays an important role in the adsorption strength, and also explains the differences in the adsorption strength of arsenic compounds under different oxidation and charge states. Moreover, we found a clear linear relation between the adsorption energies (Eads) and the electron bond density at the bond critical point of the Si–O bond (ρSi–O1) (Fig. 6), where the adsorption strength of the different methylarsenicals can be sorted as: (anionic pentavalent methylarsenicals) ≫ (neutral pentavalent methylarsenicals) > (neutral trivalent methylarsenicals), expecting that this relation could be extrapolated for a wide class of arsenic based pollutants adsorbed onto SiG.
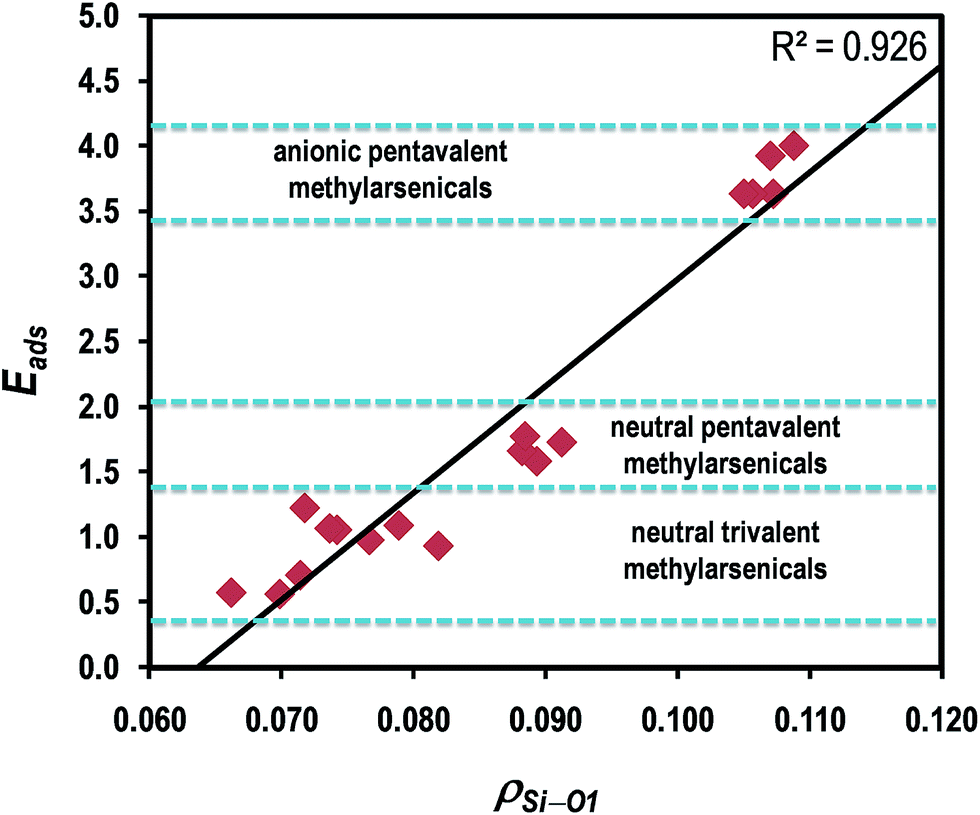 | ||
| Fig. 6 Linear relationship between the adsorption energies (Eads) and electron density at the BCP of the Si–O bond (ρSi–O) for the adsorption of methylarsenicals onto SiG. | ||
Additionally, it is noteworthy to mention that weak bond critical points were found between hydrogen atoms (belonging to the –OH or –CH3 groups of the pollutants) and carbon atoms near to the dopant in the SiG surface; these BCPs were labeled as ρC⋯H and ρC⋯H′ in Table 3, and their values remains under ∼0.02 a.u. as expected for weak closed-shell interactions.66 The graphs of the bond paths connecting the nuclear and bond critical points clearly indicate a weak C⋯H interaction that would to have a charge-controlled nature (Fig. 7a). The latter was confirmed from the plotting of the electron density difference (Fig. 7b), where is depicted the electron density distribution after the pollutant chemisorption. We can observe the attraction between the carbon atoms in SiG that are gaining electron density (sky-blue) and the pollutant hydrogen atoms losing electron density (blue). Therefore, the pollutant adsorption activates some carbon atoms of SiG, and these atoms electrostatically interact with the electron deficient hydrogen atoms of the pollutant, increasing the adsorbent–adsorbate stability.
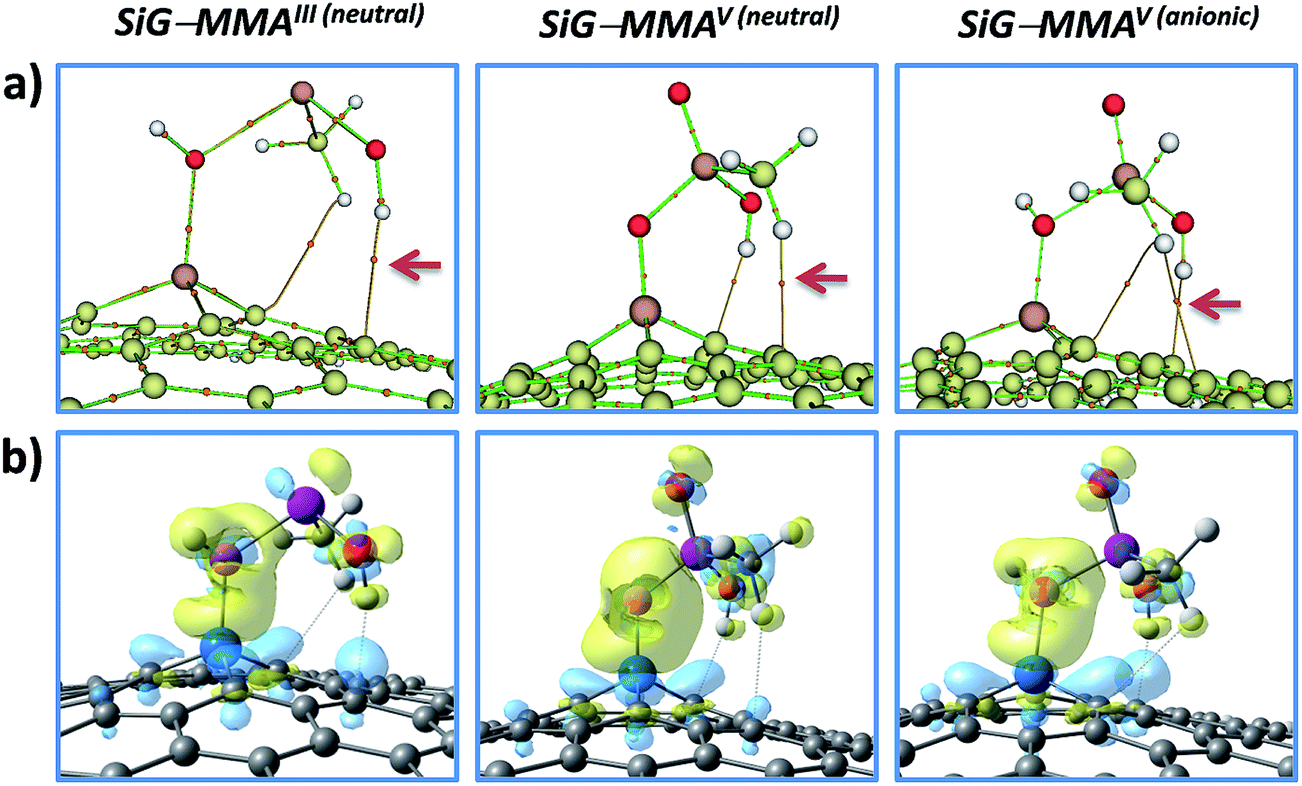 | ||
| Fig. 7 (a) Bond path connecting nuclear and bond critical points; the paths connecting the carbon (SiG) and hydrogen (pollutant) atoms suggest an electrostatic interaction between these atoms (see arrows). (b) Electron density difference; the electron density decreasing (yellow color) and electron density increasing (sky-blue color) sites are displayed after the adsorbent–adsorbate interaction. MMAIII and MMAV adsorbed in “a” conformations were taken as representatives. | ||
3.4 Solvent affects
To account for an efficient arsenic removal in water environments, it is necessary to determine if the mobility of the arsenic compounds remains low by including solvation effects; in this regard, the removal efficiency of trivalent arsenicals in aqueous environments is lower than for the pentavalent forms.59 As noted in Table 1, the H2O molecules are chemisorbed onto SiG with an adsorption energy of 0.46 eV per molecule, with a bond length of dSi–O = 2.07 Å, which is stronger that the adsorption of H2O molecules onto intrinsic graphene (<0.10 eV per molecule). Despite the latter, since the pollutant chemisorption is stronger than for H2O molecules, SiG would serve as a superior adsorbent material for the removal of inorganic and methylated arsenic compounds. To give a theoretical basis to our last point, and to gain insights into the structural effects of water molecules onto the SiG–pollutant systems, explicit/implicit solvent calculations were done to by surrounding the adsorbent–adsorbate systems with a cluster of 15H2O molecules in presence of the COSMO solvent model to create the “solvent environment”; then, the system where fully optimized in the ORCA 3.0 program.As observed in Fig. 8, the water molecules surround the adsorbed pollutants, without to cause the desorption of the compounds. Additionally, the geometrical parameters of the adsorbent–adsorbate conformations are slightly affected with respect to those determined in the gas phase (see Table S1 of the ESI†); all the Si–O bond lengths appear on the range of 1.69–1.84 Å, indicating the high stability of the chemisorption. As expected for trivalent methylarsenicals (MMAIII and DMAIII), the As–O bonds are found on the range of 2.12–2.37 Å, which agrees with the destabilization of the σAs–O bonding. Conversely, in the pentavalent methylarsenicals (MMAV, DMAV and DMMTAV), the As–O bond distances appear in the range of 1.74–1.85 Å, indicating that these compounds must show a lower mobility from the adsorbent surface compared with the trivalent forms; similar results were also obtained for iAsV. In the case of DMMTAV, the interaction by the silicon–sulfur binding is also stable in water.
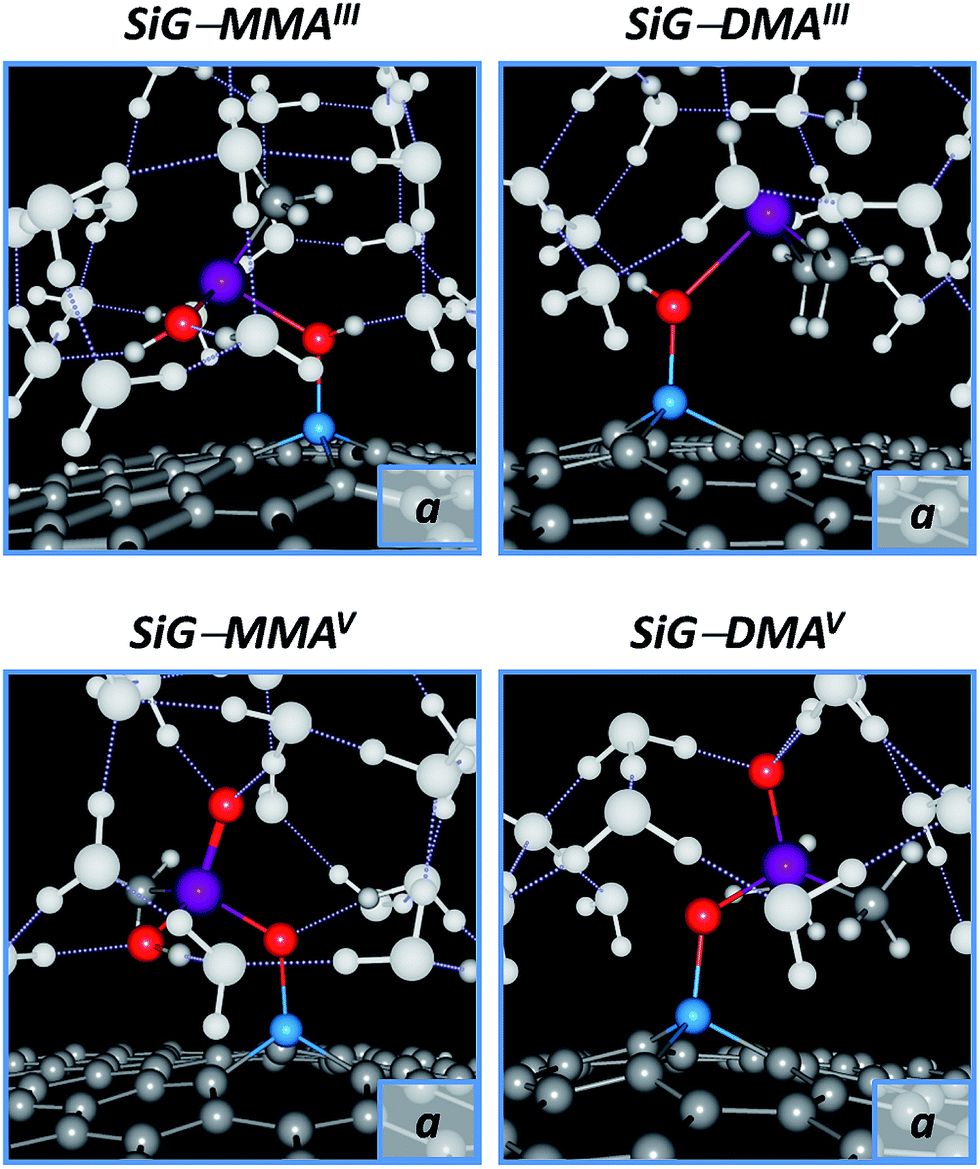 | ||
| Fig. 8 Trivalent and pentavalent methylarsenicals adsorbed onto SiG in an explicit water environment (15H2O); conformations “a” were taken as representatives. H2O molecules are depicted in white with pointed hydrogen bonds. All the systems are included in the ESI.† | ||
Additionally, it was obtained that the trivalent methylarsenicals remain protonated when they are adsorbed onto SiG, where the bond length of the interacting O–H group remains lower than ∼1.04 Å. On the other hand, in the pentavalent arsenicals, the H atom turns labile from the interacting O–H group, with the bond lengths above 1.79 Å, and forming H3O+ molecules; the latter indicates that pentavalent arsenicals are mainly adsorbed onto SiG in their anionic forms at neutral pH conditions.
3.5 Adsorption stability
In order to account for the stability of the adsorbent–adsorbate interactions, ab initio molecular dynamics trajectories were obtained at 300 K; due to the high computational cost, these calculations were performed only for the most stable systems. We studied the stability of the Si–O1 and As–O1 bonds by means of the radial pair distribution of distances [gab(r)] (Fig. 9), which allows to obtain the distribution of bond lengths along the overall trajectory; 10000 conformations per system were used for statistics.
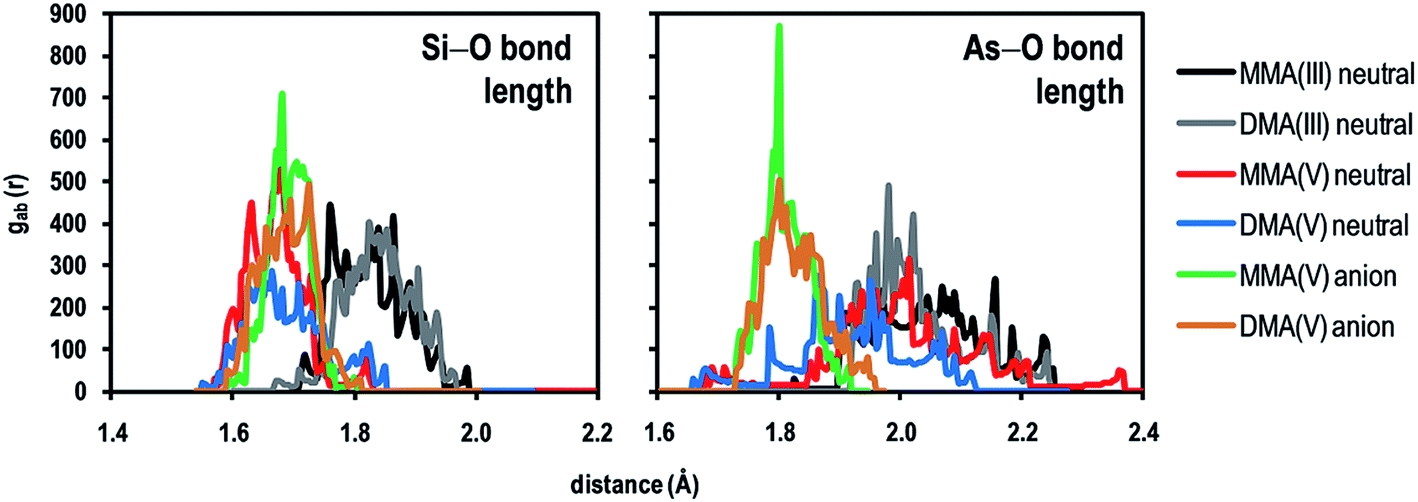 | ||
| Fig. 9 Radial pair distribution [gab(r)] of the Si–O1 and As–O1 distances in selected SiG–pollutant systems. 10000 conformations per system were used for statistics, during a time t = 1.0 ps and a temperature of 300 K. | ||
As noted in Fig. 9, the chemisorption still remains strong even in dynamic conditions, because of the Si–O1 and As–O1 bonds lengths are comparable to values obtained in the ground state. In the case of trivalent methylarsenicals (MMAIII and DMAIII), the Si–O1 and As–O1 bonds lengths are mainly retained in the range of dSi–O1 = 1.75–1.95 Å and dAs–O1 = 1.90–2.2 Å; note that although the Si–O bond is destabilized in these cases as noted above, the bond is able to be retained. Likewise, for pentavalent methylarsenicals (MMAV and DMAV), the neutral and anionic forms were analyzed; in both cases the Si–O interaction is strong and ranging from dSi–O1 = 1.69 to 1.80 Å, in agreement with the higher adsorption energies compared to the trivalent cases. However, the main differences are observed in the propagation of the As–O bonds, where the As–O bond length ranges from dAs–O1 = 1.70 to 1.90 Å for the anionic forms; while, the As–O bonds length is mainly retained in the range of dAs–O1 = 1.85–2.20 Å for the neutral forms. The latter agrees with the larger strength of the interaction of anionic arsenicals compared to its neutral forms, where differences are only given by the stability of the interacting As–O bond.
4. Conclusions
We have studied the applicability of SiG for the removal of methylarsenicals in their pentavalent and trivalent oxidation states; also comparisons with inorganic arsenic were performed. In all the cases, chemisorption is reached with high adsorption strength. In this regard, MMAIII and DMAIII are adsorbed onto SiG with adsorption energies ranging from 0.9 to 1.1 eV at neutral conditions. In the case of anionic pentavalent arsenicals (including MMAV, DMAV, DMMTAV and iAsV), the chemisorption was reached with adsorption energies in the range of 3.3–4.0 eV; while, the adsorption of their neutral forms was reached with values of up to 1.7 eV. Therefore, the adsorption strength of the different methylarsenicals adsorbed onto SiG is sorted as: (anionic pentavalent methylarsenicals) ≫ (neutral pentavalent methylarsenicals) > (neutral trivalent methylarsenicals). In addition, the adsorption strength of neutral arsenicals onto SiG was sorted as DMAV > MMAV > iAsV > DMAIII ≈ MMAIII ≈ iAsIII. We also found that the stability of the σAs–O or πAsO bonds of the methylarsenicals plays an important role in the adsorption strength, and also explains the differences in the adsorption strength of arsenic compounds under different oxidation and charge states. Additionally, from molecular dynamics and explicit solvent simulations, it was determined that the adsorption stability remains stronger both in aqueous environments and ambient conditions at 300 K. In these cases, we determined that trivalent and pentavalent methylarsenicals will be adsorbed mainly in their neutral and anionic forms at neutral pH, respectively.
Therefore, SiG is proposed as an emergent material for technologies related to the removal of organic (and inorganic) arsenic compounds in their trivalent and pentavalent oxidation states, with an enhanced performance for removal in the gas phase, and in a broad range of pH for the removal from water sources.
Acknowledgements
This work was supported by the projects FONDECYT/Postdoctorado no. 3140314, FONDECYT no. 1130072 and ICM grant no.120082.References
- W. H. Organization, Guidelines for drinking-water quality: recommendations, World Health Organization, 2004 Search PubMed
.
- W. H. O. R. O. f. Europe and W. H. Organization, Air quality guidelines: global update 2005: particulate matter, ozone, nitrogen dioxide, and sulfur dioxide, World Health Organization, 2006 Search PubMed
- V. K. Sharma and M. Sohn, Environ. Int., 2009, 35, 743–759 CrossRef CAS PubMed
- J. E. Laine, K. A. Bailey, M. Rubio-Andrade, A. F. Olshan, L. Smeester, Z. Drobná, A. H. Herring, M. Stýblo, G. G. García-Vargas and R. C. Fry, Environ. Health Perspect., 2015, 123, 186 CrossRef PubMed
- L. Yang, Y. Chai, J. Yu, B. Wei, Y. Xia, K. Wu, J. Gao, Z. Guo and N. Cui, Environ. Toxicol., 2015 DOI:10.1002/tox.22209
- D. Melak, C. Ferreccio, D. Kalman, R. Parra, J. Acevedo, L. Pérez, S. Cortés, A. H. Smith, Y. Yuan, J. Liaw and C. Steinmaus, Toxicol. Appl. Pharmacol., 2014, 274, 225–231 CrossRef CAS PubMed
- B. A. Peters, M. N. Hall, X. Liu, V. Slavkovich, V. Ilievski, S. Alam, A. B. Siddique, T. Islam, J. H. Graziano and M. V. Gamble, Environ. Res., 2015, 143, 123–130 CrossRef CAS PubMed
- E. Dopp, L. Hartmann, U. Von Recklinghausen, A. Florea, S. Rabieh, U. Zimmermann, B. Shokouhi, S. Yadav, A. Hirner and A. Rettenmeier, Toxicol. Sci., 2005, 87, 46–56 CrossRef CAS PubMed
- E. Dopp, L. Hartmann, A.-M. Florea, U. Von Recklinghausen, R. Pieper, B. Shokouhi, A. Rettenmeier, A. Hirner and G. Obe, Toxicol. Appl. Pharmacol., 2004, 201, 156–165 CrossRef CAS PubMed
- K. T. Kitchin, Toxicol. Appl. Pharmacol., 2001, 172, 249–261 CrossRef CAS PubMed
- E. M. Kenyon and M. F. Hughes, Toxicology, 2001, 160, 227–236 CrossRef CAS PubMed
- S. Wang, H. Sun, H.-M. Ang and M. Tadé, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS
- Y. Cao and X. Li, Adsorption, 2014, 20, 713–727 CrossRef CAS
- G. Z. Kyzas, E. A. Deliyanni and K. A. Matis, J. Chem. Technol. Biotechnol., 2014, 89, 196–205 CrossRef CAS
- D. Cortes-Arriagada and A. Toro-Labbe, Phys. Chem. Chem. Phys., 2015, 17, 12056–12064 RSC
- X.-L. Wu, L. Wang, C.-L. Chen, A.-W. Xu and X.-K. Wang, J. Mater. Chem., 2011, 21, 17353–17359 RSC
- K. Zhang, V. Dwivedi, C. Chi and J. Wu, J. Hazard. Mater., 2010, 182, 162–168 CrossRef CAS PubMed
- J. Zhu, R. Sadu, S. Wei, D. H. Chen, N. Haldolaarachchige, Z. Luo, J. Gomes, D. P. Young and Z. Guo, ECS J. Solid State Sci. Technol., 2012, 1, M1–M5 CrossRef CAS
- D. Nandi, K. Gupta, A. K. Ghosh, A. De, S. Banerjee and U. C. Ghosh, in Nanotechnology for Sustainable Development, Springer, 2014, pp. 149–162 Search PubMed
- G. Sheng, Y. Li, X. Yang, X. Ren, S. Yang, J. Hu and X. Wang, RSC Adv., 2012, 2, 12400–12407 RSC
- T. Wen, X. Wu, X. Tan, X. Wang and A. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 3304–3311 CAS
- S. F. Rastegar, A. A. Peyghan and N. L. Hadipour, Appl. Surf. Sci., 2013, 265, 412–417 CrossRef CAS
- Y. Chen, B. Gao, J.-X. Zhao, Q.-H. Cai and H.-G. Fu, J. Mol. Model., 2012, 18, 2043–2054 CrossRef CAS PubMed
- Y. Zou, F. Li, Z. Zhu, M. Zhao, X. Xu and X. Su, Eur. Phys. J. B, 2011, 81, 475–479 CrossRef CAS
- M. D. Esrafili, R. Nurazar and E. Vessally, Int. J. Quantum Chem., 2015, 115, 1153–1160 CrossRef CAS
- Y. Chen, X.-c. Yang, Y.-j. Liu, J.-x. Zhao, Q.-h. Cai and X.-z. Wang, J. Mol. Graphics Modell., 2013, 39, 126–132 CrossRef CAS PubMed
- R. Gholizadeh and Y.-X. Yu, Appl. Surf. Sci., 2015, 357, 1187–1195 CrossRef CAS
- J. H. Cho, S. J. Yang, K. Lee and C. R. Park, Int. J. Hydrogen Energy, 2011, 36, 12286–12295 CrossRef CAS
- R. Lv, M. C. dos Santos, C. Antonelli, S. Feng, K. Fujisawa, A. Berkdemir, R. Cruz-Silva, A. L. Elías, N. Perea-Lopez and F. López-Urías, Adv. Mater., 2014, 26, 7593–7599 CrossRef CAS PubMed
- Z. Wang, P. Li, Y. Chen, J. Liu, W. Zhang, Z. Guo, M. Dong and Y. Li, J. Mater. Chem. C, 2015, 3, 6301–6306 RSC
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533 CrossRef CAS
- M. Blanchard, K. Wright, J. D. Gale and C. R. A. Catlow, J. Phys. Chem. C, 2007, 111, 11390–11396 CAS
- Z. Wei, K. Liang, Y. Wu, Y. Zou, J. Zuo, D. C. Arriagada, Z. Pan and G. Hu, J. Colloid Interface Sci., 2016, 462, 252–259 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699–709 CrossRef CAS
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS
- H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 114, 9758–9763 CrossRef CAS
- S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 115, 10291–10302 CrossRef CAS
- H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2002, 117, 8694–8704 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Rev B01, Gaussian Inc., Wallingford CT, 2010 Search PubMed
- A. Ladeira, V. Ciminelli, H. Duarte, M. Alves and A. Ramos, Geochim. Cosmochim. Acta, 2001, 65, 1211–1217 CrossRef CAS
- G. Morin, G. Ona-Nguema, Y. Wang, N. Menguy, F. Juillot, O. Proux, F. Guyot, G. Calas and G. E. Brown Jr, Environ. Sci. Technol., 2008, 42, 2361–2366 CrossRef CAS PubMed
- A. F. Oliveira, A. C. Q. Ladeira, V. S. Ciminelli, T. Heine and H. A. Duarte, J. Mol. Struct.: THEOCHEM, 2006, 762, 17–23 CrossRef CAS
- Z. Wei, S. Zhang, Z. Pan and Y. Liu, Appl. Surf. Sci., 2011, 258, 1192–1198 CrossRef CAS
- H. Mukai and Y. Ambe, Atmos. Environ., 1987, 21(1967), 185–189 CrossRef CAS
- T. Tziaras, S. A. Pergantis and E. G. Stephanou, Environ. Sci. Technol., 2015, 49, 11640–11648 CrossRef CAS PubMed
- D. Sánchez-Rodas, A. M. S. de la Campa, D. Jesús, V. Oliveira, J. L. Gómez-Ariza, X. Querol and A. Alastuey, Chemosphere, 2007, 66, 1485–1493 CrossRef PubMed
- S. Goldberg and C. T. Johnston, J. Colloid Interface Sci., 2001, 234, 204–216 CrossRef CAS PubMed
- C. Cox and M. Ghosh, Water Res., 1994, 28, 1181–1188 CrossRef CAS
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 CrossRef CAS
- S. Suzuki, L. L. Arnold, K. L. Pennington, B. Chen, H. Naranmandura, X. C. Le and S. M. Cohen, Toxicol. Appl. Pharmacol., 2010, 244, 99–105 CrossRef CAS PubMed
- R. Raml, A. Rumpler, W. Goessler, M. Vahter, L. Li, T. Ochi and K. A. Francesconi, Toxicol. Appl. Pharmacol., 2007, 222, 374–380 CrossRef CAS PubMed
- T. Ochi, K. Kita, T. Suzuki, A. Rumpler, W. Goessler and K. A. Francesconi, Toxicol. Appl. Pharmacol., 2008, 228, 59–67 CrossRef CAS PubMed
- A. Raab, S. H. Wright, M. Jaspars, A. A. Meharg and J. Feldmann, Angew. Chem., Int. Ed., 2007, 46, 2594–2597 CrossRef CAS PubMed
- D. Mohan and C. U. Pittman, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed
- J. F. Ferguson and J. Gavis, Water Res., 1972, 6, 1259–1274 CrossRef CAS
- M. Zhang, G. He and G. Pan, Chemosphere, 2015, 122, 199–205 CrossRef CAS PubMed
- G. He, M. Zhang and G. Pan, J. Phys. Chem. C, 2009, 113, 21679–21686 CAS
- D. M. Sherman and S. R. Randall, Geochim. Cosmochim. Acta, 2003, 67, 4223–4230 CrossRef CAS
- A. Adamescu, I. Hamilton and H. A. Al-Abadleh, Environ. Sci. Technol., 2011, 45, 10438–10444 CrossRef CAS PubMed
- I. Shimoyama and Y. Baba, Carbon, 2016, 98, 115–125 CrossRef CAS
- C. F. Matta and R. J. Boyd, in The Quantum Theory of Atoms in Molecules, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 1–34 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03813a |
| This journal is © The Royal Society of Chemistry 2016 |