
Fast and reversible functionalization of a single nanopore based on layer-by-layer polyelectrolyte self-assembly for tuning current rectification and designing sensors†
Abstract
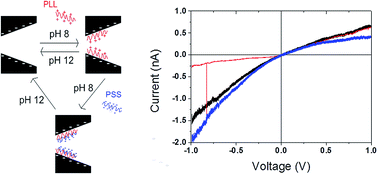
Single track-etched nanopores offer an opportunity to develop different nano-devices such as, ionic diodes or biosensors. Here we report an original approach to functionalize conical nanopores based on layer-by-layer self-assembly of poly-L-lysine and poly(styrene sulfonate). Our results show the possibility to modulate the current rectification properties and to reduce the tip diameter of the nanopore. This strategy permits a fast (less than 1 minute) and reversible functionalization of the nanopore. Based on this, we demonstrate applications in order to detect differences in polyanion (poly(styrene sulfonate), chondroitin sulfate and ssDNA) and avidin using PLL grafted with specific functions (mPEG-biotin).
 Please wait while we load your content...
Please wait while we load your content...

