
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distinguishing binding modes of a new phosphonium dye with DNA by surface-enhanced Raman spectroscopy†
Snežana
Miljanić
*a,
Adriana
Kenđel
a,
Morana
Novak
a,
Todor G.
Deliqeorqiev
b,
Ivo
Crnolatac
c,
Ivo
Piantanida
c and
Vasile
Chiş
d
aDivision of Analytical Chemistry, Department of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, HR-10000 Zagreb, Croatia. E-mail: miljanic@chem.pmf.hr; Fax: +385 1 4606 181; Tel: +385 1 4606 202
bDepartment of Pharmaceutical and Applied Organic Chemistry, Faculty of Chemistry, University of Sofia, J. Bourchier Avenue, 1164 Sofia, Bulgaria
cDivision of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, PO Box 180, HR-10002 Zagreb, Croatia
dFaculty of Physics, Babeş-Bolyai University, Kogălniceanu 1, RO-400084 Cluj-Napoca, Romania
First published on 27th April 2016
Abstract
To understand the disruption of the cell processes induced by small molecule binding to DNA, numerous structural studies of DNA complexes have been done, whereby a variety of methods has been applied. The use of various techniques is required, not only for the unique information on the structure provided by each of the methods, but also for limitations that arise during the sample preparation or measurement procedures. Here, surface-enhanced Raman scattering (SERS) spectroscopy has been employed to study binding of a new mitochondria-specific dye with ds-DNA polynucleotides. Concentration dependent differences in the SERS spectra of the dye were attributed to concentration induced changes in position of the dye molecules on the enhancing metal surface and assigned to aggregated molecules formed due to π-stacking interactions at higher dye concentration (5.0 × 10−5 mol L−1) and to single molecules at lower dye concentration (5.0 × 10−6 mol L−1). The characteristic SERS spectra were further on correlated with the spectra of the dye/DNA complexes, implying binding of the monomeric dye molecules at the excess of the adenine–thymine polynucleotide, [dye]/[AT] = 1/10, and in form of the stacked dye molecules at the higher [dye]/[AT] ratio of 1/2. In addition, the distinctive SERS spectra of the complexes with the AT alternating and homo-polymer indicated different placement of the dye molecules within the minor groove, affected by the polynucleotide groove width. Beside well-known quantitative, this study emphasized structural sensitivity of SERS, able to distinguish various molecular forms of the small molecules in the complex structures such as those formed with biomacromolecules.
Introduction
A majority of clinically important drugs are small organic molecules which non-covalently interact with DNA. While targeting nucleic acids, these molecules disrupt cell processes either by modulating gene expression or by interfering with replication.1 In order to design novel drugs or improved ones, it is necessary to understand the underlying mechanisms of the drug–DNA binding, including structural details of the formed complexes. Numerous studies imply that a detailed description of the small molecule–DNA interactions can only be unraveled by combining the information provided by different approaches and methods.There is a variety of methods devoted to structurally evidence small molecule–DNA interactions.1,2 A method for assessment of the precise binding geometry of a drug molecule relative to DNA at atomic resolution in the solid state is X-ray crystallography. Using a simplified model consisting of the drug molecule and a respective oligonucleotide, it elucidates a detailed three-dimensional molecular structure of the biological macromolecule with the embedded small molecule; yet, it is often limited by the sample preparation step, i.e. crystallization of the drug–DNA complex. To reveal the structure of the drug–DNA complex in solution, which is more relevant to physiological processes, NMR spectroscopy has been extensively used. Despite the emphasis on the flexible nature of the DNA molecule, difficulties here arise from the size of the biomolecule, since the increase in the number of the signals with molecular weight complicates interpretation of the observed spectra. Apart from providing undoubtedly valuable data on the structure of the small molecule–DNA complexes in different environments, both methods, X-ray crystallography and NMR spectroscopy, are considered as highly demanding in respect of time, experience, and expense. Additionally, simpler methods of electronic spectroscopy (absorption, fluorescence, circular dichroism (CD) spectroscopy) have been widely applied for small molecule–DNA studies in solution, allowing the use of long DNA/RNA polynucleotides. Though changes in optical properties of the chromophore moieties imply binding modes of the small molecules with DNA, these methods lack ability to provide more detailed structural data, not indicating the exact binding sites of the interacting molecules. Information on the molecular structure based on characteristic spectral patterns as molecular “fingerprints” can be observed by means of vibrational methods, infrared (IR) and Raman spectroscopy. Spectral changes correlated to the structures of the interacting molecules before and after binding allow identification of the reactive sites of the species involved. Owing to water ability to strongly absorb IR radiation and poorly scatter radiation, Raman spectroscopy is the more convenient method for monitoring molecular interactions in aqueous systems, commonly used to mimic cell media. Both vibrational methods, however, suffer from low sensitivity, resulting in use of large amounts of (bio)chemicals that makes these methods rather expensive. Therefore, new, sensitive and cost-effective methods are emerging, such as surface-enhanced Raman scattering (SERS) spectroscopy.
SERS spectroscopy is a powerful vibrational spectroscopy technique that allows for highly sensitive structural detection of low concentration molecules in vicinity to nanostructured metal surfaces.3 Owing to recent developments in design and fabrication of plasmonic substrates, essential for the Raman scattering enhancement, application of SERS spectroscopy in analytical, biophysical and life sciences constantly increases.4 The enhancement of the Raman scattering is attributed to two mechanisms: an electromagnetic mechanism and a chemical mechanism. The electromagnetic mechanism occurs for the molecules adsorbed on the metallic nanostructures due to the amplification of electromagnetic field arising from the excitation of the surface plasmons. The chemical mechanism, on the other hand, implies chemisorption of the molecule onto the metal surface, allowing transfer of electrons from the metal to the molecule and vice versa. Even though the charge transfer mechanism is considered to contribute to the overall enhancement to a lesser extent than the electromagnetic one, the total SERS enhancement factor with respect to the normal Raman signal, in most cases is the product of both mechanisms, reaching up to 10.10
Owing to the detailed structural data, the high sensitivity and the fluorescence quenching, the SERS technique seems to be a well-suited method for biomolecular recognition studies. Besides, numerous practical advantages make the SERS spectroscopy the method of choice in DNA targeting research: (i) measurements are performed in micromolar concentration range, thus avoiding difficulties with the substances of poor solubility and aggregation tendency. (ii) Neither the sample preparation nor the data interpretation are limited by the size of the biomolecule, allowing structural analysis of complexes with basepair sequences of various length, oligonucleotides as well as polynucleotides. (iii) Using colloidal suspensions as the SERS active substrates, the measurements are performed in aqueous medium at physiological pH, mimicking the cell conditions and allowing flexibility of DNA. (iv) Due to the high sensitivity of the technique (bio)chemicals are used in small quantities, resulting in low cost measurements. (v) The sample is simply prepared by mixing solutions of the small molecule and DNA with the colloid.
However, an application of the SERS spectroscopy in revealing drug–DNA interactions has been mostly overlooked, resulting in a very few papers published on complexes with long polynucleotides. Given the two principal modes of the small molecule–DNA non-covalent binding, majority of the SERS studies aimed at intercalation,5–13 while study of groove binding by SERS was almost neglected.14–16 Besides, our studies on small molecule–DNA binding using SERS spectroscopy,17–22 not only implied binding modes in agreement with those proposed by other applied structural methods, but also complemented already observed results with new information on structural moieties involved in interactions.
Recently, a series of sterically restricted phosphonium dyes were synthesized and their ability to interact with DNA/RNA polynucleotides studied in detail using methods of electronic spectroscopy (UV/Vis absorbance, fluorescence and CD) and molecular modelling.23,24 For the phosphonium dye containing pyridinium oxazole, quinoline and triphenylphosphonium moiety in the structure (Scheme 1), CD spectra indicated concentration dependent groove-binding with DNA, particularly interesting and distinctive of adenine–thymine polynucleotides.23 It was suggested that at excess of AT polynucleotide (r[dye]/[AT] < 1/5) the phosphonium dye binds as monomer, while at high molar ratio (r[dye]/[AT] > 1/5) dye molecules form a dimer within the polynucleotide minor groove. In order to obtain structural data about the formed dye/AT complexes, SERS spectroscopy was applied. In addition, to provide a reliable and accurate assignment and interpretation of the obtained spectra computational studies were performed.
 | ||
| Scheme 1 Molecular structure of the phosphonium dye. | ||
Experimental
Chemicals and solutions
Silver nitrate (Kemika) and trisodium citrate (Kemika) were of analytical reagent grade and used as supplied for the silver colloid preparation. Water was purified by passage through Milli-Q (Millipore) deionizing and filtration columns. Cacodylate buffer contained 0.05 mol L−1 sodium cacodylate adjusted to pH 7.0 by 0.1 mol L−1 HCl. Polynucleotides, consisting of 500–800 base pairs, (polydA–poly dT, polydAdT–polydAdT and polydGdC–polydGdC) were purchased from Sigma-Aldrich and dissolved in the cacodylate buffer, I = 0.05 mol L−1, pH = 7.0. Concentration of polynucleotides: c(polydA–polydT) = 1.00 × 10−3 mol L−1, c(polydAdT–polydAdT) = 7.35 × 10−3 mol L−1, c(polydGdC–polydGdC) = 1.00 × 10−4 mol L−1, was determined spectroscopically as the concentration of phosphates. A detailed synthesis and spectroscopic characterization of the dye, 4-methyl-2{[1-(3-(triphenylphosphonio)-propyl)quinolin-4(1H)-ylidene]methyl}oxazolo[4,5-b]pyridinium diiodide has been published elsewhere.24 Stock solution of the dye, c = 2.00 × 10−4 mol L−1, was prepared by dissolving the substance in Milli-Q water.Colloid preparation
Before preparation of colloid all glassware was thoroughly cleaned with a detergent solution, followed by treatment in 5% nitric acid, and finally rinsed with water of Milli-Q purity. Silver colloid was prepared according to the modified Lee and Meisel reduction method with citrate.25,26 Silver nitrate (18 mg) was dissolved in water (100 mL) and heated rapidly to boiling under stirring. A 1% (w/v) trisodium citrate aqueous solution (2 mL) was added to the boiling solution and kept gently boiling for 90 min with continuous stirring. The resulting colloidal solution was greenish yellow with the maximum at 418 nm in the UV/Vis/NIR absorption spectrum. A pH value of the prepared colloid was 7.8.Samples preparation
For the concentration dependent SERS measurements samples were prepared by mixing the silver colloidal solution and an appropriate volume of the dye stock solution (2.00 × 10−4 mol L−1). The final concentrations of the dye were in range between 1.0 × 10−6 and 5.0 × 10−5 mol L−1. Interactions with polynucleotides were studied in the silver colloid containing the dye, c = 5.0 × 10−6 mol L−1, and a respective double stranded polynucleotide in [dye]/[polynucleotide] ratios of 1/2 and 1/10. All working samples were prepared by mixing the dye solution with the polynucleotide solution, followed by addition of the silver colloid. In case of time dependent SERS measurements, and in order to avoid influence of the silver nanoparticles on formation of the dye/polynucleotide complexes, the colloid was added 2 and 50 minutes after mixing the dye and the polynucleotide. The total volume of all the samples was 500 μL, containing the same amount of the silver colloid (375 μL).For the UV/Vis/NIR absorption measurements samples were prepared in the same way as for the SERS measurements. However, in order to obtain a measurable value of the silver colloid absorbance the samples were diluted with Milli-Q water in a sample/water volume ratio of 1/2 prior to the measurement.
Instrumentation
FT-Raman and SERS spectra were measured on a Bruker Equinox 55 interferometer equipped with a FRA 106/S Raman module. Nd-YAG laser excitation at 1064 nm and a laser power of 100 and 500 mW were used for the measurement of the solid substance and colloidal solutions, respectively. The Raman spectrum was acquired from the solid dye on an aluminum holder, while glass vials were used for handling colloidal samples. Spectra were taken in the 3500–100 cm−1 range. To obtain a good spectral definition, 128 scans at a spectral resolution of 4 cm−1 were averaged for a spectrum. In addition, due to poor reproducibility of the SERS spectra usually obtained using colloidal suspensions as the SERS active substrates, the spectra were acquired for three colloidal samples of each measured system. When spectral features in three measured spectra resembled each other and were affected only by the spectral noise, the spectra were considered sufficiently reproducible and the averaged spectrum was further on analyzed and interpreted (see ESI, Fig. S1†).UV/Vis/NIR absorption measurement of the silver colloid and colloidal samples was carried out with an Analytik Jena spectrometer (model SPECORD 200). Conventional quartz cells (10 mm × 10 mm) were used throughout.
For pH measurements, a Mettler Toledo pH meter (model MP 220) with a Mettler Toledo InLab®413 combined glass-calomel electrode was used. The pH meter was calibrated with standard aqueous buffer solutions of pH 7.00 and 4.01.
Computational methods
For geometry optimizations and Raman spectra calculations the hybrid B3LYP exchange–correlation functional in conjunction with Pople's 6-31G(d) (BS1) and 6-311G(d,p) (BS2) basis sets was used.27–31Because of the lack of structural data for the investigated phosphonium dye, its conformational landscape was initially explored in vacuo at B3LYP/BS1 level of theory. After finding the most stable conformer, its geometry and vibrational properties were refined using the more flexible basis set BS2, both, in vacuo and water. The implicit Polarizable Continuum Model (PCM) was used to account for solvent effects.32
For the calculations performed on dimers the BS1 basis set for H, C, N, O and P atoms and the cc-pVTZ basis set and SDB effective core potential for iodide atoms were used according to Marten et al.,33 as a good compromise between accuracy and computational costs for such large molecules.
Frequency calculations confirmed that all the optimized geometries correspond to true minima on the potential energy surfaces. Prior to compare the calculated harmonic wavenumbers with the experimental values, the former have been scaled using the scaling factor f = 0.967, excepting the high-wavenumbers corresponding to X–H stretchings for which a scaling factor of 0.955 was applied.34
The calculated Raman activities were converted to relative Raman intensities according to Williams et al.35 For plotting the calculated Raman spectra, pure Lorentzian band shapes were used with the half-width at half-maximum (HWHM) of 7 cm−1, as derived from the experimental data. The assignment of the experimental bands was based on the observed frequencies and intensity patterns of the experimental spectra and confirmed by establishing a one-to-one correlation between the experimental and theoretical calculated frequencies. Visual inspection of modes was also used for a proper assignment of spectra, by considering both the band positions and intensity patterns.
NMR spectra of phosphonium dye conformers, both for dication and neutral forms, were calculated applying the GIAO (Gauge-Including Atomic Orbital) method,36,37 at B3LYP/pc-2//B3LYP/pc-2 level of theory.38 In order to express the chemical shifts in terms of the total computed NMR shielding tensors, the standard referencing method to tetramethylsilane (TMS) was used. For this purpose, the geometry of TMS molecule (Td symmetry) had been optimized and then its NMR shielding tensors were obtained at the same level of theory as that used for the dye.
All the calculations were performed with the Gaussian09 software package.39 Figures representing the structures of the phosphonium dye were created using the Mercury 3.3 program.40
Results and discussion
Conformational analysis
Optimization of the molecular structure of the phosphonium dye was performed considering initially the two conformers with N-methyl group and quinoline moiety in cis (c1) and trans positions (c2), respectively (Scheme 2). At B3LYP/BS1 level of theory, c2 is more stable than c1 by 5.22 kcal mol−1. Next, the conformations of c2 differing in the rotations around the rotatable bonds in the propyl chain were explored. All the other conformers are less stable than c2 with relative energies between 0.15 and 4.09 kcal mol−1 (ESI, Table S1†). | ||
| Scheme 2 B3LYP/6-311G(d,p) optimized geometries of c1 and c2 conformers in gas-phase. | ||
The relative stability of c1 and c2 conformers is preserved also at the B3LYP/BS2 level of theory. Thus, for dications, c2 is more stable than c1 by 5.33 and 7.72 kcal mol−1 in gas-phase and water, respectively. For the neutral species in water, c2 is again more stable by 4.31 kcal mol−1. In DMSO, at B3LYP/pc-2 level of theory, c1 is even more destabilized than c2, by 8.13 kcal mol−1.
The greater stability of the c2 conformer in solution is also supported by NMR data. Thus, the calculated chemical shifts for this conformer are in much better agreement with the experimental data in DMSO reported by Tumir et al.23 Particularly, the predicted chemical shift for H53 for the c1 conformer is 11.46 ppm at B3LYP/pc-2 level of theory, a value which is in an unacceptable discrepancy with the reported experimental data (7.35–8.54 ppm).23 Moreover, NMR data also support the presence of the dicationic species in solution (ESI, Table S2†). As easily can be observed, in case of neutral c2 conformer, the proton chemical shifts are much worse reproduced as for dication, particularly for methyl, methylene and methine protons.
Besides the c1 and c2 conformers, two kinds of dimers consisting of two monomers in c2 conformation, oriented with the triphenylphosphonium groups on the same and opposite sides, respectively, were also optimized (ESI, Scheme S1†). Four iodide anions were necessary to stabilize the charge of the dimers. The very small calculated relative energy between the two dimers (0.05 kcal mol−1) points to the fact that in solution they coexist. In Fig. 1, the Raman spectrum obtained as the sum of the spectra corresponding to the individual dimers is also included and the calculated wavenumbers in the cumulated spectrum are appended in Table 1.
 | ||
| Fig. 1 (a) The experimental Raman spectrum of the solid phosphonium dye. The calculated Raman spectra of: (b) the cationic c22+ conformer in gas-phase, (c) the cationic c22+ conformer in water, (d) the neutral c2 conformer in water and (e) the dimers with the phosphonium substituents oriented on the same and opposite sides in gas-phase. | ||
| Wavenumber (cm−1) | Assignments | |||||
|---|---|---|---|---|---|---|
| Experimental | Calculated | |||||
| Raman | SERS | SERS | c22+ gas-phase | c22+ water | Dimers | |
| (5.0 × 10−5 M) | (5.0 × 10−6 M) | |||||
| a Abbreviations: vw: very weak, w: weak, m: medium, s: strong, vs: very strong, sh: shoulder, ν: stretching, δ: deformation, γ: torsion, τ: twisting, ω: umbrella, s: symmetrical, as: asymmetrical, ip: in plane, oop: out-of-plane, ar: aromatic, Ox: oxazole, P: pyridinium, Ph: phenyl, Q: quinoline. | ||||||
| 3098 vw | 3090 | 3096 | 3106 | ν(CH)P | ||
| 3059 w | 3057 w | 3057 w | 3063 | 3063 | 3073 | ν(CH)Ph |
| 2949 vw | 2926 vw | 2931 | 2952 | 2941 | ν s(CH3) + νs(CH2) | |
| 2893 vw | 2899 vw | 2905 | 2925 | 2931 | ν s(CH2) | |
| 2861 vw | 2872 vw | 2870 vw | 2840 | ν s(CH2) | ||
| 1655 w | 1656 w | 1656 m | 1633 | 1634 | 1637 | ν(CC)P |
| 1625 vvw | 1615 vvw br | 1621 m | 1613 | ν(CC)Q + ν(CC)Ox–CH | ||
| 1609 w | 1609 vw | 1610 sh | 1606 | 1602 | 1610 | ν(CC)Q |
| 1588 w | 1587 vw | 1587 w | 1570 | 1572 | 1586 | ν(CC)Ph + ν(CC/CN)P,Ox,Q |
| 1537 m | 1538 s | 1539 m | 1528 | 1533 | 1532 | [ν(CC) + δ(CH)]Ox,Q,P |
| 1508 w | 1508 | 1514 | 1510 | ν(Q) + ν(CC)Q-CH | ||
| 1483 m | 1477 m | 1480 m | 1476 | δ ip(P,Ox,Q) + δas(CH3) + δ(CH2) | ||
| 1470 s | 1471 | 1466 | δ ip(P,Ox) + δas(CH3) | |||
| 1453 m | 1452 m | 1454 m | 1458 | 1430 | 1457 | δ ip(P,Ox) + ω(CH3) + δ(CH2) |
| 1422 vs | 1426 s | 1425 s | 1432 | 1412 | 1436 | ν(CN)P,Ox + ν(CC)Q + δ(CH) + δas(CH3) |
| 1408 sh | 1411 s | 1415 sh | 1402 | 1399 | 1399 | ν(CC,CN)P,Ox + δ(CH) + ω(CH3) |
| 1370 w | 1363 | 1362 | 1365 | δ(CH2) + ν(CN)Q | ||
| 1350 s | 1351 vs | 1352 s | 1324 | 1324 | 1334 | [ν(CC) + ν(CN)]Q + δ(CH) + ω(CH2) |
| 1328 sh | 1319 w | 1300 | 1301 | 1329 | δ ip(P,Ox,Q) + ρ(CH3) + δip(CH) | |
| 1285 w | 1277 vw | 1281 w | 1264 | 1263 | 1265 | [ν(CC) + ν(CN)]Q,Ox,P |
| 1248 m | 1251 w | 1250 w | 1236 | 1224 | 1229 | ν(CO) + δip(Q,Ox,P) |
| 1219 w | 1219 | 1211 | δ ip(CH)Q + δ(CH2) | |||
| 1206 w | 1189 | 1194 | 1187 | δ ip(Q) + δip(CH)Q + τ(CH2) | ||
| 1177 vw | 1163 | 1164 | δ ip(CH)P + ν(N–CH3) | |||
| 1154 w | 1152 w | 1133 | 1137 | 1140 | δ ip(Q) + τ(CH2) + δip(CH)Q | |
| 1107 vw | 1105 w | 1106 w | 1090 | 1088 | 1116 | δ ip(P,Ox) + ρ(CH3) |
| 1026 w | 1029 m | 1029 m | 1010 | 1011 | 1010 | δ ip(Ph) |
| 1001 m | 1000 s | 1000 s | 982 | 982 | 973 | δ ip(Ph) + ν(CH2–CH2) |
| 893 w | 895 w | 894 w | 909 | 906 | 908 | δ ip(Ox,P) |
| 834 w | 843 | 846 | 842 | δ ip(Q) + δ(CH2) | ||
| 803 w | 797 m | 790 m | 812 | 811 | 804 | δ ip(Q) |
| 727 vw | 725 vw | 725 w | 734 | 725 | δ ip(P,Ox,Q,Ph) | |
| 686 vw | 684 w | 678 m | 699 | 698 | δ ip(P,Ox,Q) | |
| 625 w | 628 w | 628 w | 640 | 639 | 638 | δ ip(P,Ox,Q) |
| 596 m | 596 m | 597 m | 608 | 608 | 610 | δ ip(P,Ox,Q) |
| 564 vw | 567 w | 569 w | 567 | 574 | 587 | δ oop(Q) |
| 540 w | 540 sh | |||||
| 535 w | 525 s | 526 s | 535 | 537 | 539 | δ ip(Q) + δ(CPC) + γ(Ph) |
| 513 vw | 522 | 522 | δ ip(Q) + δ(CPC) + γ(Ph) | |||
| 494 w | 494 w | 494 w | 501 | 501 | 501 | δ ip(P,Q) + δ(CPC) |
| 471 vw | 468 w | 470 w | 479 | 479 | 481 | δ ip(Q,P,Ox) |
| 238 m | 260 m | 258 m | 240 | 233 | 241 | δ(CPC) |
| 224 m | 220 sh | |||||
| 199 w | 199 | 188 | 191 | δ(CPC) + δoop(Q) | ||
| 174 m | 174 | 174 | 183 | δ(Q–CH–Ox) | ||
Raman spectrum of the phosphonium dye
The experimental Raman spectrum of the dye, together with the calculated spectra for dicationic form in gas phase, the calculated spectra of both, neutral and dicationic species in water and the cumulated spectrum of the two dimers are shown in Fig. 1. The assignments of the most intense bands are given in Table 1 and they are based on the agreement between the calculated and experimental data, both for band position and their relative intensities pattern. It is worth mentioning that the band at 2861 cm−1 can only be reproduced by using the dimer model.In the Raman spectrum of the crystalline dye weak bands at 3098 and 3059 cm−1 were assigned to C–H stretchings in aromatic moieties, whereas very weak bands between 2950 and 2860 cm−1 were attributed to asymmetric and symmetric C–H stretchings of the methyl and methylene groups (Fig. 1 and Table 1). A pattern of intense bands in the spectral range 1655−1200 cm−1 originated from vibrational modes of the highly conjugated part of the molecule consisting of the pyridinium (P), oxazole (Ox) and quinoline (Q) rings (Scheme 2 and Table 1). It was dominated by a very strong band at 1422 cm−1, associated with mixed vibrations of the aromatic moieties, and accompanied by medium to intense bands at 1537, 1470 and 1350 cm−1. Stretching of the pyridinium ring gave rise to the weak band at 1655 cm−1, while deformation of the pyridinium oxazole system contributed to the strong band at 1470 cm−1. Deformations of the phenyl rings in triphenylphosphonium substituent produced bands of moderate intensity at 1026 and 1001 cm−1. Bands in the wavenumber range lower than 1000 cm−1 resulted mostly from deformation and torsion modes of the aromatic units, including weak bands at 535, 494 and 238 cm−1 to which also contributed vibrational modes of the CPC group containing the phosphorus atom.
Due to poor solubility of the substance in water, the Raman spectrum of the phosphonium dye in solution was not obtained, i.e. the spectrum of the 2 × 10−4 M aqueous dye solution corresponded to the Raman spectrum of water.
SERS spectra
 | ||
| Fig. 2 UV/Vis/NIR absorption spectra of the silver colloid (black), the silver colloid containing the phosphonium dye (red) and the dye/polynucleotide complexes: [dye]/[polydA–polydT] = 1/2 (blue), [dye]/[polydA–polydT] = 1/10 (cyan), [dye]/[polydGdC–polydGdC] = 1/2 (purple), [dye]/[polydGdC–polydGdC] = 1/10 (magenta). Given that samples were diluted with water to obtain measurable absorbance of the colloid, the final concentration of the dye was 1.7 × 10−6 mol L−1. | ||
The addition of the dye/polynucleotide complexes into the colloidal suspension affected the absorption spectrum to a lesser extent. Nevertheless, a shift towards 419 nm, slight decrease in intensity and broadening of the silver plasmon band indicated changes onto or near the enhancing metallic surface. Shoulders at 526 nm and 529 nm appeared, implying presence of the dye/polynucleotide complexes in a ratio of 1/2 and 1/10, respectively,23 while bands at 255 and 257 nm were attributed to the guanine–cytosine and adenine–thymine polynucleotide in the complex containing the higher amount of the polynucleotide (r[dye]/[poly] = 1/10). Owing to the excess of the polynucleotide in the complexes, regardless of the dye/polynucleotide ratio, the negative charge of the formed complexes prevented efficient adsorption on the negatively charged silver nanoparticles, finally resulting in poorer aggregation.
Nevertheless, the broad band at longer wavelengths, obtained for the silver nanoparticle aggregates with the phosphonium dye, allowed SERS measurements of the studied systems using NIR excitation.
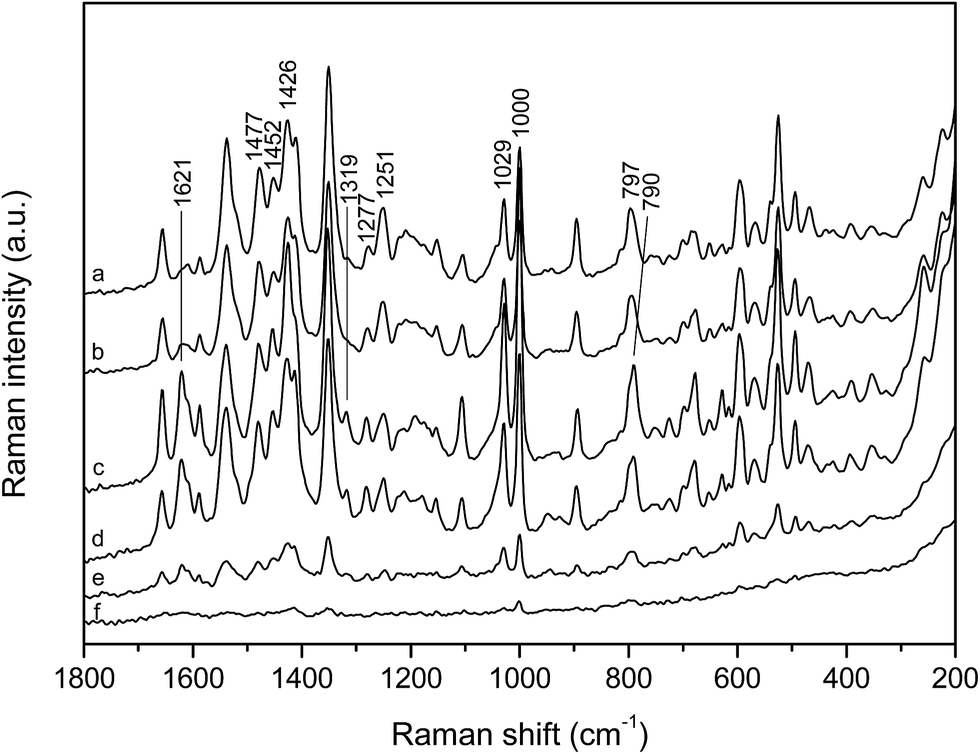 | ||
| Fig. 3 SERS spectra of the phosphonium dye in the silver colloid at various concentrations: (a) 5.0 × 10−5 mol L−1, (b) 1.0 × 10−5 mol L−1, (c) 5.0 × 10−6 mol L−1, (d) 2.5 × 10−6 mol L−1, (e) 1.5 × 10−6 mol L−1 and (f) 1.0 × 10−6 mol L−1. Excitation at 1064 nm. The spectra are displaced for visual clarity. | ||
According to the selection rules on the enhancing metallic surface the most enhanced scattering originates from vibrations with polarizability change perpendicular to the surface, while the change in polarizability parallel to the surface does not contribute to the scattering.3 Given that SERS spectra result from the surface selection rules, they are indicative of orientation of the dye molecules on the enhancing surface, which was in this case obviously concentration dependent, further pointing to molecular forms present on the silver nanoparticles. An existence of molecular aggregates on the silver surface had been already implied by SERS in our previous study with ethidium bromide.17
The SERS spectra indicated that the phosphonium dye molecules were densely packed on the silver nanoparticles at high concentration (5.0 × 10−5 mol L−1), when intermolecular stacking between the aromatic pyridinium oxazole and quinoline moieties took place (Scheme 3a). By lowering concentration the number of the molecules on the metal nanoparticles decreased, allowing single molecules to adopt the optimal orientation towards the silver surface while forming a molecular monolayer (Scheme 3b). A considerable change in relative band intensities, accompanied by the shift of the quinoline ring deformation band, implied weakening of the π-stacking interactions, leading to the selectively enhanced vibrational modes of the single molecules. Besides, the appearance of the new bands (1621 and 1319 cm−1) pointed to the placement of the single dye molecules on the silver surface clearly different from that attained in the form of aggregates. Having in mind negative charge of the silver nanoparticles, provided by the stabilizing layer of the citrate anions on their surface, the dye molecules were most likely adsorbed through the methylpyridinium cation and/or triphenylphosphonium cation due to attractive electrostatic interactions. Indeed, the B3LYP/BS2 theoretical charge pattern for c2 dication supported this assumption, the calculated NBO charges being +0.72e, −0.14e and −0.414e for pyridinium, oxazole and quinoline moieties, respectively, while a partial charge of +1.6e was predicted for the phosphorus atom. On the other hand, the charge pattern was drastically different for the neutral c2 conformer in water, particularly for the quinoline moiety. The calculated NBO charges in this case were +0.44e, −0.122e, −0.939e and +1.596e for P, Ox and Q moieties and P atom, respectively. The new bands in the low concentration dye SERS spectra, associated with the pyridinium oxazole and quinoline ring vibrations as well as with the N-methyl group rocking (1319 cm−1), implied an almost perpendicular position of the conjugated aromatic system of the single molecules with the methylpyridinium cation close to the enhancing surface, rather than with the bulky triphenylphosphonium cation.
 | ||
| Scheme 3 A schematic view of the proposed orientation of the phosphonium dye molecules on the silver nanoparticle surface at concentration: (a) 5.0 × 10−5 mol L−1 and (b) 5.0 × 10−6 mol L−1. | ||
Finally, it was proposed that the spectral signature of the dye at the highest measured concentration, 5.0 × 10−5 mol L−1, corresponded to the molecular aggregates, while the SERS spectrum at the dye concentration of 5.0 × 10−6 mol L−1 was distinctive of the monomeric molecules.
To monitor time dependent reorientation of the dye molecules on the silver surface, SERS spectra of the phosphonium dye at concentrations of 5.0 × 10−5 and 5.0 × 10−6 mol L−1 were measured during one hour. The identical SERS spectra of the dye at 5.0 × 10−5 mol L−1, acquired immediately after sample preparation and 50 minutes later, indicated that densely packed molecular structures formed on the metal nanoparticles did not change their position with time (ESI, Fig. S2†). However, an increase in intensity of the monomer characteristic bands (1621 and 1319 cm−1), obtained after 50 minutes in the spectrum of the dye at 5.0 × 10−6 mol L−1, implied that the single molecules, unlike the stacked ones, freely reoriented. Hence the quinoline and pyridinium oxazole moieties took more upright position on the silver surface, with the side consisting of nitrogen atoms close to it. The reorientation was most likely driven by attractive electrostatic interactions between the methylpyridinium cation and anionic surface layer on the enhancing silver nanoparticles.
By the NIR excitation (1064 nm) of silver colloids containing polynucleotides alone, SERS spectra were not observed. The acquired spectra resembled the spectrum of the pure colloid (mainly water), except for the broad citrate band at 1092 cm−1, which was missing in the spectra of the polynucleotides (ESI, Fig. S3†). Taking into account the fact that colloidal nanoparticles carried negative charge, the highly negatively charged phosphate backbone of long polynucleotides most likely prevented efficient adsorption onto the enhancing metal surface. Yet, being able to displace citrate ions on the metal surface,41,42 polynucleotides were presumably placed close to the silver nanoparticles. However, given the surface plasmon resonance of the silver colloid in the visible region and the dependence of the Raman intensity, i.e. sensitivity, on the laser radiation frequency (ν4), a lack of the polynucleotides SERS spectra was attributed to 1064 nm excitation. In addition, in order to avoid presence of any substances, which were not involved in dye/polynucleotide complex formation, colloidal samples were not aggregated with sulfate salts, successfully used for label free DNA detection.41,42
The scattered radiation of the complexes with both AT polynucleotides in the studied molar ratios (r[dye]/[AT] = 1/2 and 1/10) was of very weak intensity, indicating binding of the dye with the AT rich biomolecules (Fig. 4c–f). The reduced intensity was a consequence of a distant position of the formed complex from the silver surface, caused by repulsive forces between the stabilizing anionic layer and negatively charged polynucleotides. Nevertheless, a doublet of the phenyl bands at 1028 and 1000 cm−1, observed in all the SERS spectra with the adenine–thymine polynucleotides implied position of the dye triphenylphosphonium substituent in vicinity to the enhancing metal surface, thus outside the AT helix (Scheme 4).
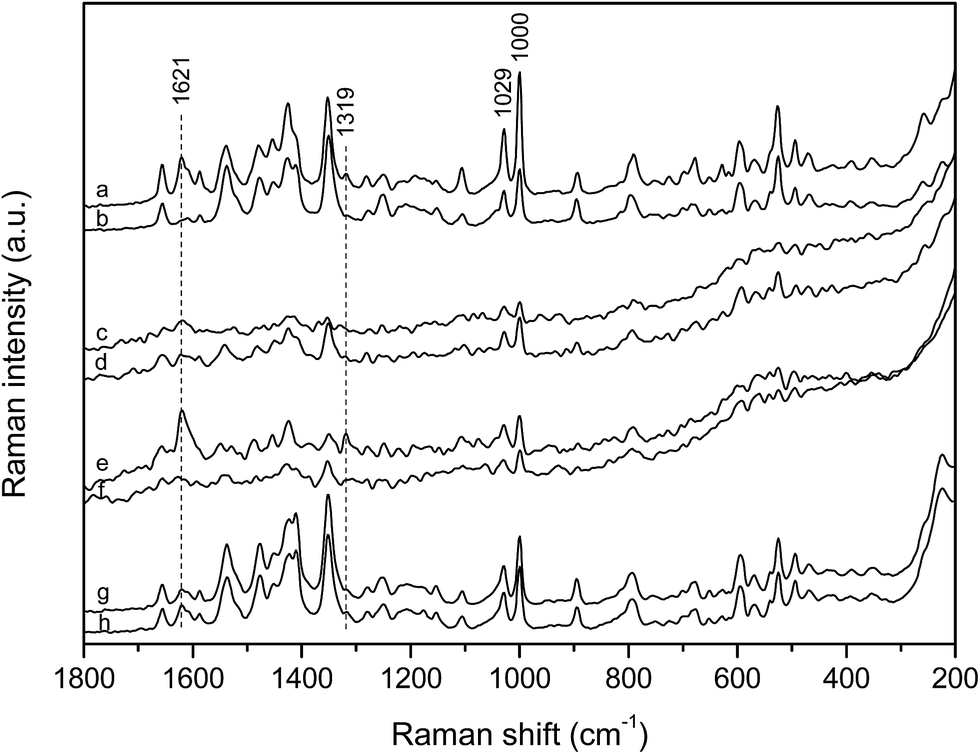 | ||
| Fig. 4 SERS spectra of: (a) phosphonium dye monomers, 5.0 × 10−6 mol L−1, and (b) phosphonium dye aggregates, 5.0 × 10−5 mol L−1, in comparison to the SERS spectra of the phosphonium dye/polynucleotide complexes: (c) [dye]/[polydA–polydT] = 1/10, (d) [dye]/[polydA–polydT] = 1/2, (e) [dye]/[polydAdT–polydAdT] = 1/10, (f) [dye]/[polydAdT–polydAdT] = 1/2, (g) [dye]/[polydGdC–polydGdC] = 1/10 and (h) [dye]/[polydGdC–polydGdC] = 1/2; c(dye)complex = 5.0 × 10−6 mol L−1. Excitation at 1064 nm. The spectra are displaced for visual clarity. The SERS spectra of the complexes with the AT polynucleotides are magnified by 5 times. | ||
 | ||
| Scheme 4 A schematic view of the proposed binding modes of the phosphonium dye with AT polynucleotides with respect to the silver nanoparticle surface: (a) [dye]/[polydAdT–polydAdT] 1/10, (b) [dye]/[polydAdT–polydAdT] 1/2; (c) [dye]/[polydA–polydT] 1/10; (d) [dye]/[polydA–polydT] 1/2. | ||
At the polynucleotide excess, r[dye]/[AT] = 1/10, the marker band of the monomeric molecules at 1621 cm−1 was clearly obtained (Fig. 4c and e), being more pronounced and accompanied by another monomer characteristic band at 1319 cm−1 in the spectrum of the complex with polydAdT–polydAdT (Fig. 4e). This spectral pattern implied that the dye molecules accommodated as monomers into the minor groove of the AT polynucleotides. In addition, the wider minor groove of the alternating AT polynucleotide (6.3 Å), if compared to the minor groove of the AT homo-polymer (3.3 Å), allowed monomeric molecules to more easily access the enhancing silver surface, resulting in stronger enhancement of the scattered radiation, unlike the poor scattering of the dye molecules placed deep in the minor groove of polydA–polydT (Scheme 4a and c).
At the [dye]/[AT] 1/2 ratio the spectral features resembled more those associated with the molecular aggregates (Fig. 4d and f), as expected upon population of all the DNA binding sites by the single molecules. The SERS spectrum of the complex with polydAdT–polydAdT was pretty noisy with the only recognizable bands of the phenyl rings at 1028 and 1000 cm−1 and of quinoline at 1351 cm−1 (Fig. 4f). Nevertheless, it supported molecular modeling data on head-to-tail orientation of the dye molecules within the groove, with the positively charged phosphonium substituents placed on the opposite sides and outside the helix (Scheme 4b and d).23 Hence the optimal groove width of the alternating AT polynucleotide enabled binding of the stacked dye molecules as dimers into the minor groove. The SERS spectrum of the dye with polydA–polydT (Fig. 4d), on the other hand, implied coexistence of the monomeric and associated molecular structures within the minor groove. This was in agreement with the proposed slow kinetics of the dimer formation caused by the narrow minor groove of the AT homo-polymer.
In order to monitor binding of the dye molecules with AT polynucleotides with time, the SERS spectra were measured 50 minutes after the complex formation. At the polynucleotide excess, r[dye]/[AT] = 1/10, when the dye was bound as monomer within the minor groove, the spectra were not changed during time (ESI, Fig. S4†). However, at the high ratio, r[dye]/[AT] = 1/2, the observed spectra, although noisy and of poor intensity, differed from those acquired immediately after sample preparation and resembled each other regardless of the polynucleotide (ESI, Fig. S4†). Since a slight broadening of the minor groove (<10%) due to dye dimer binding had been suggested,23 it was very likely that once dimers were formed within the groove, their orientation with respect to the enhancing silver surface differed from the initial one, but their position was very similar in the minor groove of both polynucleotides.
Unlike the spectra of the dye with AT polynucleotides, the SERS spectra with polydGdC–polydGdC at both measured ratios (r[dye]/[AT] = 1/2 and 1/10) were very similar and quite intense (Fig. 4g and h), indicating their origin from the free dye molecules on the silver surface rather than from the complexed ones. However, the spectral characteristics did not unambiguously correspond to either monomeric or aggregated structures, most likely resulting from the dye molecules at concentration lower than 5.0 × 10−6 mol L−1, being inclined towards the silver surface. Free dye molecules on the enhancing surface pointed to the weak binding of the dye with the GC polynucleotide, which had been implied by the circular dichroism data.23 Despite the wide minor groove (9.5 Å) of the alternating GC polynucleotide, the guanine amino group within the minor groove sterically hindered efficient binding of the studied dye molecules.
Conclusions
To obtain structural data on binding of the phosphonium dye with DNA, the surface-enhanced Raman scattering spectroscopy was applied, supported by the comprehensive theoretical study on the dye conformers as well as single and associated dye molecules. The distinctive SERS spectra allowed differentiation between the monomeric and aggregated forms of the studied dye, which were further on correlated with the molecular structures formed upon binding within the DNA minor groove. The SERS spectra implied binding of the single molecules with the adenine–thymine polynucleotides at the excess of the biopolymer (r[dye]/[AT] = 1/10), while indicated existence of the stacked molecules within the minor groove of the AT polynucleotides at the high dye/AT ratio (r[dye]/[AT] = 1/2). The observed SERS spectra of the complexes also differed depending on the polynucleotide, as a consequence of the polynucleotide structural properties. Hence it was possible to distinguish the positions of the dye molecules within the minor groove of the AT alternating polymer and homo-polymer, which were dependent on the minor groove width, as well as to suggest weak binding of the dye molecules with the GC polynucleotide due to the sterical hindrance of the guanine amino group in the minor groove. The observed SERS spectra thus provided an experimental structural confirmation of the binding modes proposed by electronic spectroscopy and molecular modelling in the previous studies.Finally, this work pointed to a huge potential of the SERS spectroscopy as a powerful technique providing adequate and reliable structural information on the small molecule–DNA binding. Moreover, in case of here presented DNA-sequence selective fluorimetric dye, the SERS spectroscopy proved to be very sensitive independent method for checking the results obtained by fluorescence as well as for checking the structural data of dye/DNA complex obtained by CD spectrophotometry.
References
- S. Neidle, Principles of Nucleic Acid Structure, Elsevier, London, 2008 Search PubMed.
- K. Nakamoto, M. Tsuboi and G. D. Strahan, Drug-DNA Interactions: Structures and Spectra, John Wiley & Sons, New Jersey, 2008 Search PubMed.
- R. Aroca, Surface-Enhanced Vibrational Spectroscopy, John Wiley & Sons, Chichester, 2006 Search PubMed.
- S. Schlücker, Surface-Enhanced Raman Spectroscopy: Analytical, Biophysical and Life Science Applications, Wiley-VCH, Weinheim, 2011 Search PubMed.
- H. Morjani, J.-F. Riou, I. Nabiev, F. Lavelle and M. Manfait, Cancer Res., 1993, 53, 4784–4790 CAS.
- I. Nabiev, I. Chourpa and M. Manfait, J. Phys. Chem., 1994, 98, 1344–1350 CrossRef CAS.
- I. Nabiev, A. Baranov, I. Chourpa, A. Beljebbar, G. D. Sockalingum and M. Manfait, J. Phys. Chem., 1995, 99, 1608–1613 CrossRef CAS.
- A. Ianoul, F. Fleury, O. Duval, R. Waigh, J.-C. Jardillier, A. J. P. Alix and I. Nabiev, J. Phys. Chem. B, 1999, 103, 2008–2013 CrossRef CAS.
- G. Breuzard, J.-M. Millot, J.-F. Riou and M. Manfait, Anal. Chem., 2003, 75, 4305–4311 CrossRef CAS PubMed.
- A. Murza, S. Alvarez-Méndez, S. Sanchez-Cortes and J. V. Garcia-Ramos, Biopolymers, 2003, 72, 174–184 CrossRef CAS PubMed.
- P. Zhao, L.-C. Xu, J.-W. Huang, B. Fu, H.-C. Yu, W.-H. Zhang, J. Chen, J.-H. Yao and L.-N. Ji, Bioorg. Chem., 2008, 36, 278–287 CrossRef CAS PubMed.
- W. Xie, Y. Ye, A. Shen, L. Zhou, Z. Lou, X. Wang and J. Hu, Vib. Spectrosc., 2008, 47, 119–123 CrossRef CAS.
- J. Wang and X. Yang, Spectrochim. Acta, Part A, 2009, 74, 421–426 CrossRef PubMed.
- M. Ermishov, A. Sukhanova, E. Kryukov, S. Grokhovsky, A. Zhuze, V. Oleinikov, J.-C. Jardillier and I. Nabiev, Biopolymers, 2000, 57, 272–281 CrossRef CAS PubMed.
- C.-Y. Wu, W.-Y. Lo, C.-R. Chiu and T.-S. Yang, J. Raman Spectrosc., 2006, 37, 799–807 CrossRef CAS.
- M. Masetti, H.-N. Xie, Ž. Krpetić, M. Recanatini, R. A. Alvarez-Puebla and L. Guerrini, J. Am. Chem. Soc., 2015, 137, 469–476 CrossRef CAS PubMed.
- S. Miljanić, A. Dijanošić, I. Matošević and I. Piantanida, Vib. Spectrosc., 2011, 57, 23–29 Search PubMed.
- S. Miljanić, A. Dijanošić, M. Kalac, M. Radić Stojković, I. Piantanida, D. Pawlica and J. Eilmes, Croat. Chem. Acta, 2012, 85, 577–584 CrossRef.
- S. Miljanić, A. Dijanošić, K. Landeka, M. Radić Stojković and I. Piantanida, Appl. Spectrosc., 2012, 66, 82–89 CrossRef.
- M. Radić Stojković, S. Miljanić, K. Mišković, L. Glavaš-Obrovac and I. Piantanida, Mol. BioSyst., 2011, 7, 1753–1765 RSC.
- S. Miljanić, A. Dijanošić, I. Piantanida, Z. Meić, M. Teresa Albelda, A. Sornosa-Ten and E. García-Espana, Analyst, 2011, 136, 3185–3193 RSC.
- A. Dijanošić, S. Miljanić, I. Piantanida, J. González-García and E. García-Espana, J. Raman Spectrosc., 2014, 45, 863–872 CrossRef.
- L.-M. Tumir, I. Crnolatac, T. Deligeorgiev, A. Vasilev, S. Kaloyanova, M. Grabar Branilović, S. Tomić and I. Piantanida, Chem.–Eur. J., 2012, 18, 3859–3864 CrossRef CAS PubMed.
- I. Crnolatac, L.-M. Tumir, N. Y. Lesev, A. A. Vasilev, T. G. Deligeorgiev, K. Mišković, L. Glavaš-Obrovac, O. Vugrek and I. Piantanida, ChemMedChem, 2013, 8, 1093–1103 CrossRef CAS PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- C. H. Munro, W. E. Smith, M. Garner, J. Clarkson and P. C. White, Langmuir, 1995, 11, 3712–3720 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilka and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. I. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- J. Marten, W. Seichter, E. Weber and U. Bohme, CrystEngComm, 2008, 10, 541–547 RSC.
- R. D. Johnson III, K. K. Irikura, R. N. Kacker and R. Kessel, J. Chem. Theory Comput., 2010, 6, 2822–2828 CrossRef PubMed.
- S. D. Williams, T. J. Johnson, T. P. Gibbons and C. L. Kitchens, Theor. Chem. Acc., 2007, 117, 283–290 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision C.01), Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Rowler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- E. Papadopoulou and S. E. J. Bell, Angew. Chem., Int. Ed., 2011, 50, 9058–9061 CrossRef CAS PubMed.
- E. Papadopoulou and S. E. J. Bell, Chem.–Eur. J., 2012, 18, 5394–5400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03586h |
| This journal is © The Royal Society of Chemistry 2016 |