
Catalytic cross-benzoin/Michael/acetalization cascade for asymmetric synthesis of trifluoromethylated γ-butyrolactones†
Abstract
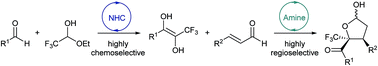
A sequential NHC-amine catalytic cascade reaction has been developed to assemble aromatic aldehydes, trifluoroacetaldehyde ethyl hemiacetal and enals asymmetrically into CF3-substituted chiral γ-butyrolactone derivatives featuring vicinal quaternary and tertiary stereocenters. This approach incorporates a highly chemoselective intermolecular cross-benzoin reaction and a highly regioselective Michael-acetalization cascade. Various multi-functionalized tetrahydrofuran scaffolds were readily prepared from hemiacetal intermediates through convenient organic transformations.
 Please wait while we load your content...
Please wait while we load your content...

