
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atmospheric sink of methyl chlorodifluoroacetate and ethyl chlorodifluoroacetate: temperature dependent rate coefficients, product distribution of their reactions with Cl atoms and CF2ClC(O)OH formation†
María B.
Blanco
a,
Ian
Barnes
b,
Peter
Wiesen
b and
Mariano A.
Teruel
*a
aCONICET, Instituto de Investigaciones en Fisicoquímica, I.N.F.I.Q.C., Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, 5000 Córdoba, Argentina. E-mail: mteruel@fcq.unc.edu.ar; Fax: +54-351-4334188
bBergische Universität Wuppertal, Fakultät für Mathematik und Naturwissenschaften, Physikalische & Theoretische Chemie, Gauss Strasse 20, 42119 Wuppertal, Germany
First published on 20th May 2016
Abstract
Rate coefficients as a function of temperature have been measured for the first time for the gas-phase reactions of chlorine atoms with methyl chlorodifluoracetate (k1) and ethyl chlorodifluoroacetate (k2) using the relative rate technique. The experiments were carried out in a 1080 L photoreactor over the temperature range 287–313 K at a total pressure of 1000 ± 10 mbar of synthetic air using in situ FTIR spectroscopy to monitor reactants and products. The following Arrhenius expressions were obtained: k(MCDFA+Cl) = (9.6 ± 5.1) × 10−12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−(1363 ± 79)/T] and k(ECDFA+Cl) = (64.4 ± 29.7) × 10−12exp[−(1110 ± 68)/T]. The kinetic results are compared with previous experimental and theoretical studies. In addition, a product study of the reactions of Cl with methyl chlorodifluoracetate and ethyl chlorodifluoroacetate is reported. The results indicate that in the absence of NOx the main fate of the alkoxy radicals formed after H-atom abstraction by Cl from the –CH3 group in methyl chlorodifluoroacetate is reaction of the radical with O2 to form the mixed anhydride CF2ClC(O)OC(O)H. In the case of ethyl chlorodifluoroacetate the main fate of the alkoxy formed via H-atom abstraction by Cl from the –CH2– entity in the ethyl group is α-ester rearrangement to produce chlorodifluoroacetic acid and the corresponding radical. The yields of chlorofluoracetic acid (CF2ClC(O)OH) obtained were as follows: (34 ± 5)% and (86 ± 8)% for the reactions of Cl with CF2ClC(O)OCH3 and CF2ClC(O)OCH2CH3, respectively. The measured yields are rationalized in terms of mechanisms consisting of competitive reaction channels for the formed products in the oxidation, i.e. reaction with O2, α-ester rearrangement and a decomposition pathway. Atmospheric implications are discussed according to the rate coefficients obtained as a function of temperature and altitude, and regarding the formation of chlorofluorocarboxylic acid.
exp[−(1363 ± 79)/T] and k(ECDFA+Cl) = (64.4 ± 29.7) × 10−12exp[−(1110 ± 68)/T]. The kinetic results are compared with previous experimental and theoretical studies. In addition, a product study of the reactions of Cl with methyl chlorodifluoracetate and ethyl chlorodifluoroacetate is reported. The results indicate that in the absence of NOx the main fate of the alkoxy radicals formed after H-atom abstraction by Cl from the –CH3 group in methyl chlorodifluoroacetate is reaction of the radical with O2 to form the mixed anhydride CF2ClC(O)OC(O)H. In the case of ethyl chlorodifluoroacetate the main fate of the alkoxy formed via H-atom abstraction by Cl from the –CH2– entity in the ethyl group is α-ester rearrangement to produce chlorodifluoroacetic acid and the corresponding radical. The yields of chlorofluoracetic acid (CF2ClC(O)OH) obtained were as follows: (34 ± 5)% and (86 ± 8)% for the reactions of Cl with CF2ClC(O)OCH3 and CF2ClC(O)OCH2CH3, respectively. The measured yields are rationalized in terms of mechanisms consisting of competitive reaction channels for the formed products in the oxidation, i.e. reaction with O2, α-ester rearrangement and a decomposition pathway. Atmospheric implications are discussed according to the rate coefficients obtained as a function of temperature and altitude, and regarding the formation of chlorofluorocarboxylic acid.
Introduction
Oxygenated Volatile Organic Compounds (OVOCs) such as hydrofluoroethers (HFEs) and hydrochlorofluoroethers (HCFEs) have an oxygen linkage which makes the hydrogen atoms of the ether molecule more labile and increases their reactivity toward OH radicals and Cl atoms in the lower atmosphere compared to chlorofluorocarbons (CFCs) and hydrochlorofluorocarbons (HCFCs). This increased atmospheric photooxidation potential of HFEs and HCFEs over CFCs and HCFCs makes them good replacement materials in many applications such as solvents, heat-transfer fluids in refrigeration systems, cleaning agents in the electronic industry, foam-blowing, lubricant deposition, and dry etching agents and as anesthetics.1,2The hydroxyl radical (OH) is the primary cleaning agent of the lower atmosphere and is thus the dominant sink for many greenhouse gases and pollutants. In coastal and marine regions and in inland areas near to pollutant sources the concentration of Cl can attain concentration levels which make reactions with Volatile Organic Compounds (VOCs) competitive with the OH-radical initiated degradation of these VOCs.3–6 In recent years, evidence has been presented for Cl-atom chemistry in continental regions far removed from coastal and marine regions where observations of Cl-atom precursors, have been made.7–10 Additionally, urban sources of Cl atoms due to industrial activities can exist, and Cl concentrations higher than the concentration levels observed in marine regions have been reported, for example, in the vicinity of ceramic industries.11
Chlorinated fluoroesters (CFESs) are emitted to the atmosphere mainly by anthropogenic sources. Methyl chlorodifluoroacetate (MCDFA) is used as a building block in organic synthesis12 and ethyl chlorodifluoroacetate (ECDFA) is employed extensively in fire retardants and also finds use in the production of pesticides, dyestuffs, phosphates and medicine.13
MCDFA and ECDFA can be produced directly in the troposphere through the photooxidation of hydrochlorofluoroethers analogous to the formation of fluoroesters from HFEs.14,15 In order to evaluate if HCFEs are environmentally acceptable replacements for CFCs and HCFCs, rate coefficients and product distributions for the reactions of the CFESs with the main tropospheric oxidants are necessary.
Hence, as part of a systematic study on the kinetics and oxidation products of the atmospheric reactions of haloesters, we report in this work, rate coefficients for the reactions of Cl atoms with methyl chlorodifluoroacetate and ethyl chlorodifluoracetate measured over the temperature range 287–313 K using a relative kinetic technique:
| ClCF2C(O)OCH3 + Cl → products | (1) |
| ClCF2C(O)OCH2CH3 + Cl → products | (2) |
This study extends our previous kinetic database for the title reactions at 298 K16 through provision of the temperature dependence for the reactions.
To the best of our knowledge, the temperature dependence of the reactions has not been previously reported. Although, there are many kinetic and mechanistic studies of the reactions of Cl atoms with a variety of hydrogenated esters, the database for haloesters is very limited. Furthermore, there is no information available on the degradation mechanisms of chlorofluorinated esters in the atmosphere and the resulting products.
Therefore, the main reaction products of the haloesters have been identified and their yields quantified. This information has been used to propose atmospheric degradation mechanisms for the Cl-atom mediated oxidation of the compounds. Formation of chlorodifluoracetic acid has been observed in high yields. Field measurements of the acid in precipitation suggest that it may accumulate in the aquatic enviroment.17
The aim of the present work is to determine the fate of the chlorofluoroalkoxy radical formed in the Cl-initiated photodegradation of the chlorofluoroacetates studied, and appraise its possible contribution to the accumulation in the environment of chlorofluorinated compounds such as chlorodifluoracetic acid (CDFA).
Experimental
The experiments were performed in a 1080 L photoreactor a detailed description of which can be found elsewhere thus only a brief general overall description is given here.18 A diagram of the reactor is presented below (Scheme 1). The reactor has a total length of 6.2 m and an internal diameter of 0.47 m, it consists of two quartz glass cylinders which are connected by a central enamel flange ring and silicon rubber seals and is closed at both ends by aluminum flanges which contain inlet ports for reactants etc. The reactor can be temperature regulated within the range 287 to 313 K with a precision of ±2 K through a regulation system using three PT-100 thermo-elements. The chamber can be evacuated by a pumping system consisting of a turbo-molecular pump backed by a double stage rotary fore pump to 10−3 Torr. The photolysis system consists of 32 superactinic fluorescent lamps (Philips TL05 40 W), which are evenly spaced around the reaction vessel. The lamps emit light with a maximum intensity at 360 nm. The chamber is equipped with a White type multiple-reflection mirror system for sensitive in situ long path absorption monitoring of reactants and products in the IR spectral range 4000–700 cm−1 and was operated at total optical path length of 484.7 ± 0.8 m. IR spectra were recorded with a spectral resolution of 1 cm−1 using a Nicolet Nexus FT-IR spectrometer equipped with a KBr beam splitter and a liquid nitrogen cooled mercury–cadmium–telluride (MCT) detector.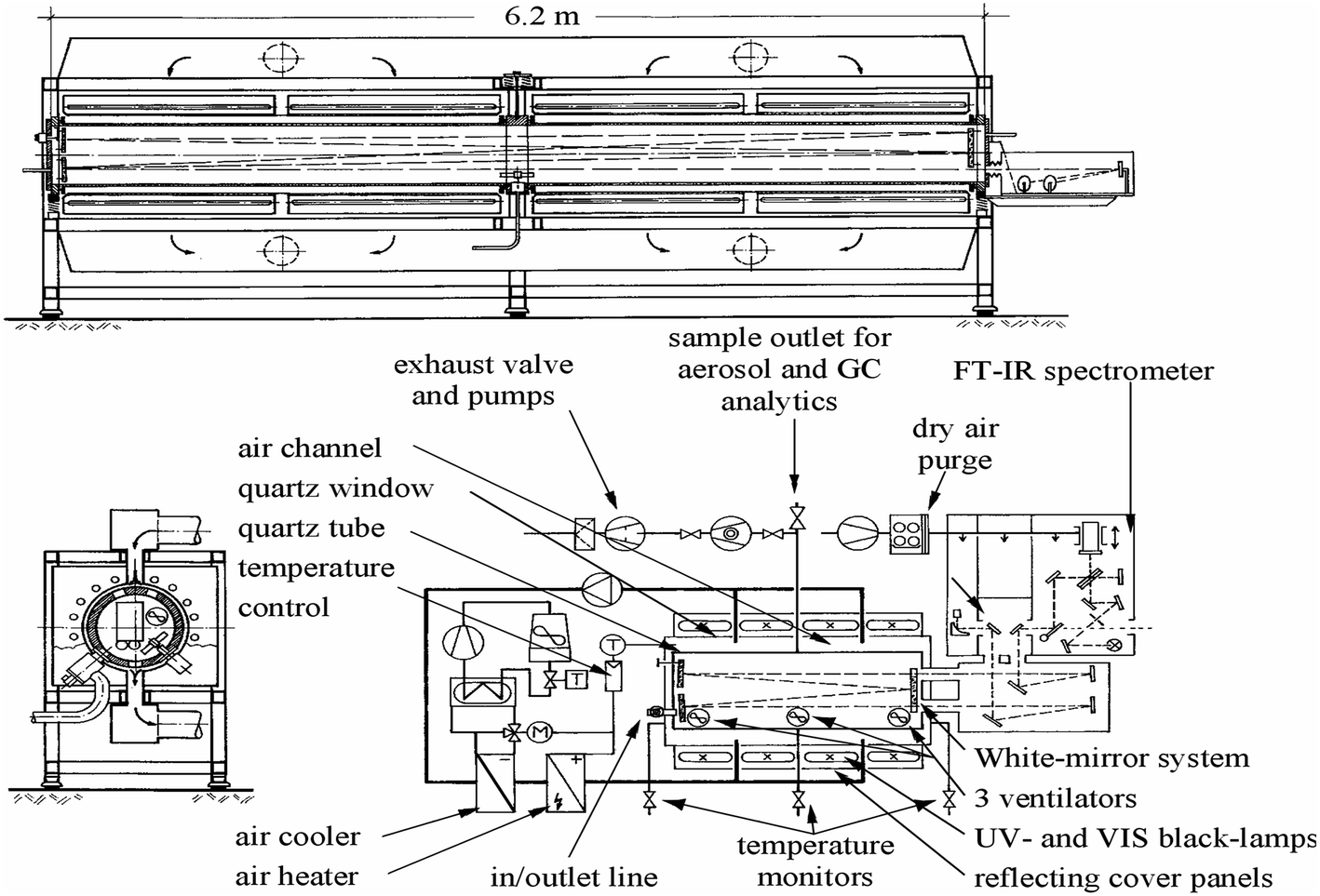 | ||
| Scheme 1 Schematic diagram of the 1080 L quartz-glass reactor. | ||
The rate coefficients for the reaction of Cl atoms with the chlorofluoroacetates were determined using the relative kinetic technique. Chlorine atoms were generated by photolysis of molecular Cl2:
| Cl2 + hν → 2Cl | (3) |
In the presence of Cl atoms, the chlorofluoroacetates and the reference compounds decay through the following reactions:
| Cl + chlorofluoroacetate → products, kchlorofluoroacetate | (4) |
| Cl + reference → products, kreference | (5) |
Provided that the reference compound and the reactant are lost only by reactions (4) and (5), then it can be shown that:
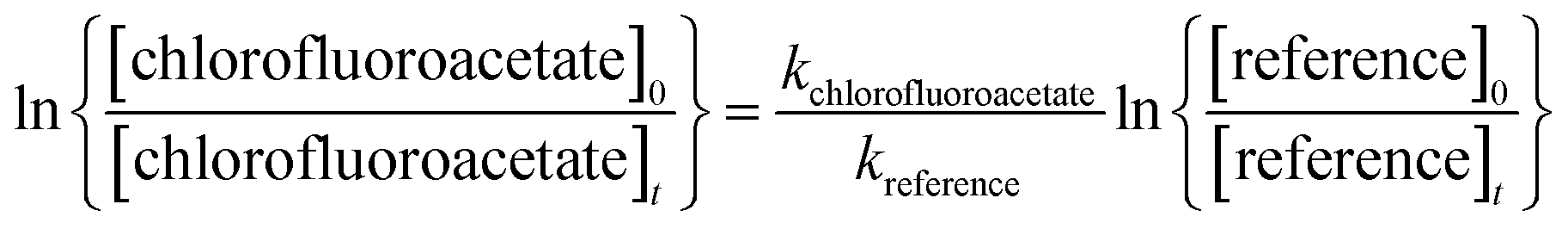 | (I) |
The relative rate technique relies on the assumption that both the chlorofluoroacetates and reference organics are removed solely by reaction with Cl atoms. To verify this assumption, mixtures of chlorine and both organics (chlorofluoroacetate and reference hydrocarbon) in air were prepared and allowed to stand in the dark for 30 minutes the typical duration of an experiment. In all cases, reaction of the organic species with the Cl-atom precursor Cl2 in the absence of UV light was of negligible over this time period. Additionally, to test for possible photolysis of the reactants mixtures of the chlorofluoroesters and reference compounds in air in the absence of molecular chlorine were irradiated using the output from all the fluorescent lamps surroundings the chamber for 30 min. Photolysis was not observed for any of the reactants.
For the kinetic investigations chloromethane was used as the reference compound and experiments were performed on Cl2/chlorofluoroester/chloromethane/air mixtures which were irradiated for periods of 15–20 minutes during the course of which infrared spectra were recorded with the FTIR spectrometer. Typically 64 interferograms were co-added per spectrum over a period of approximately 1 minute. Similar experiments were performed for the product studies without the reference compound in the reaction mixtures.
Results and discussion
Temperature dependence of the reactions of Cl atoms with MCDFA and ECDFA
Rate coefficients for the reactions of Cl atoms with MCDFA and ECDFA were determined at atmospheric pressure over the temperature range of 287–313 K relative to the reaction of Cl atoms with chloromethane. Fig. S1 and S2† show examples of the kinetic data obtained at three temperatures (287, 303 and 313 K) for both methyl chlorodifluoroacetate and ethyl chlorodifluoroacetate plotted according to eqn (I). The rate coefficient ratios kCFES/kreference obtained from linear least squares analyses of such plots are listed in Table 1. The rate coefficient ratios are each from a minimum of at least three or four experiments for each temperature and CFES and the errors are the 2σ statistical errors from the linear regression analyses.| T (K) | k CFES/kreference | k CFES × 1013 (cm3 per molecule per s) | CFES | E a/R (K) | A × 1012 (cm3 per molecule per s) |
|---|---|---|---|---|---|
| Methyl chlorodifluoroacetate | |||||
| 287 | 0.19 ± 0.01 | 0.83 ± 0.36 | |||
| 294 | 0.20 ± 0.02 | 0.93 ± 0.41 | |||
| 298 | 0.20 ± 0.01 | 0.98 ± 0.44 | 1363 ± 79 | 9.6 ± 5.1 | |
| 303 | 0.20 ± 0.02 | 1.12 ± 0.49 | |||
| 313 | 0.21 ± 0.02 | 1.24 ± 0.52 | |||
| 287 | 3.14 ± 0.05 | 13.53 ± 4.36 | Ethyl chlorodifluoroacetate | ||
| 294 | 3.08 ± 0.04 | 14.29 ± 4.38 | |||
| 298 | 3.13 ± 0.04 | 15.59 ± 4.43 | 1110 ± 68 | 64.4 ± 29.7 | |
| 303 | 3.12 ± 0.03 | 17.10 ± 4.52 | |||
| 313 | 3.06 ± 0.04 | 18.64 ± 4.69 |
The rate coefficients for the reaction of Cl atoms with the chlorofluoroacetates listed in Table 1 have been put on an absolute basis using rate coefficients for Cl with chloromethane at the respective temperatures calculated from the following Arrhenius expression (with k in cm3 per molecule per s units):
| k(Cl + chloromethane) = 3.3 × 10−11e−(1250)/T cm3 per molecule per s.19 |
The errors given for the rate coefficients are a combination of the 2σ statistical errors from the linear regression analyses plus an added uncertainty to take into account the error associated with the values of the reference rate coefficients.
The values of the rate coefficients for the reactions of Cl atoms with methyl chlorodifluoroacetate and ethyl chlorodifluoroacetate as a function of temperature are plotted in Arrhenius form in Fig. 1. Linear least-squares analysis of the plots leads to the following Arrhenius expressions in units of cm3 per molecule per s:
| k(MCDFA+Cl) = (9.6 ± 5.1) × 10−12exp[−(1363 ± 79)/T] |
| k(ECDFA+Cl) = (64.4 ± 29.7) × 10−12exp[−(1110 ± 68)/T] |
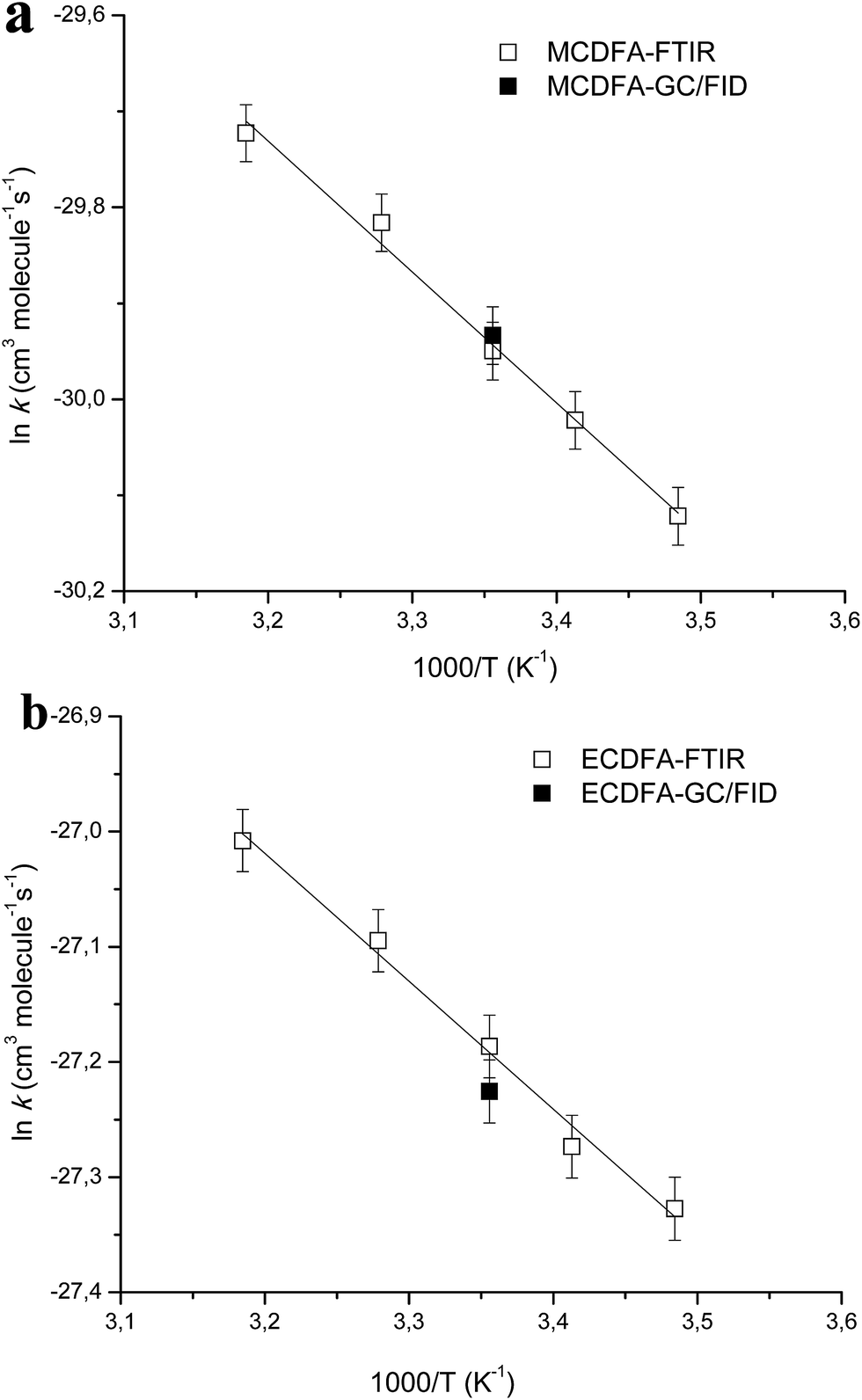 | ||
| Fig. 1 Arrhenius plots of the temperature dependent kinetic data obtained for the reactions of Cl atoms with MCDFA (a) and ECDFA (b) in the present study. For comparison, the room temperature rate coefficient from our previous work16 is also shown (filled symbol). | ||
The errors in the activation term and the pre-exponential factor are the 2σ random statistical errors from fits to the data presented in Table 1 and plotted in Fig. 1 (panels a and b). To the best of our knowledge there are no prior experimental determinations of the Arrhenius parameters for these reactions, hence, this study represents the first temperature dependence study of the reactions of Cl atoms with MCDFA and ECDFA.
The positive Ea/R values of (1363 ± 79) K and (1110 ± 68) K for the reaction of Cl atoms with MCDFA and ECDFA, respectively, indicates the dominance of a hydrogen abstraction pathway under the experimental conditions i.e. the rate coefficients increase slightly with temperature.
The Ea/R values of 1363 K and 1110 K reported in this work for the reactions of MCDFA + Cl and ECDFA + Cl, respectively are consistent with the available Ea/R literature data for the reactions of Cl atoms with hydrofluoroethers and fluoroesters where Ea/R values of 852, 1190, 2254, 968, 1216, 1637 and 1368 K have been reported for the reactions of Cl atoms with HFE-7200 (C4F9OC2H5), HFE-7500 (n-C3F7CF(OC2H5)CF(CF3)2), HFE-7100 (C4F9OCH3), HFE-7300 (n-C2F5CF(OCH3)CF(CF3)2), CF3C(O)OCH3, CF3C(O)OCH2CH3 and CF2HC(O)OCH3, respectively.20–23 The employment of temperature dependent kinetic data in model calculations increases the accuracy of the models since the temperature gradient of the troposphere and the seasonal temperature variations can be taken into consideration.
Comparison with previous kinetic determinations
The following rate coefficients have been obtained at 298 K for the reaction of Cl with MCDFA and ECDFA:| k1 = (0.98 ± 0.44) × 10−13 cm3 per molecule per s |
| k2 = (15.55 ± 4.43) × 10−13 cm3 per molecule per s |
The values of k1 and k2 at room temperature are in very good agreement with our previously reported values16 of k(Cl+MCDFA) = (1.0 ± 0.2) × 10−13 and k(Cl+ECDFA) = (15 ± 3) × 10−13 cm3 per molecule per s16 obtained at 298 K and 750 Torr total pressure in a 80 liter Teflon bag using the relative kinetic method with gas chromatography/flame ionization detection (RR-GC/FID) for the analyses. These two measurements are represented in Fig. 1 by filled symbols, for the reaction of Cl with MCDFA (Fig. 1a) and ECDFA (Fig. 1b), respectively. Mishra et al.24 have calculated the rate coefficient for H-atom abstraction from CF2ClC(O)OCH3 by Cl atoms using canonical transition state theory (CTST) and employing Wigner25 and Eckart26 symmetrical methods which gave values of 0.96 × 10−13 and 1.20 × 10−13 cm3 per molecule per s, respectively, at 298 K for the reaction. These values are in very good agreement with the experimental value of 0.98 × 10−13 cm3 per molecule per s reported in this work and the previous experimental value reported by our group, using a different experimental setup, of (1.0 ± 0.2) × 10−13 cm3 per molecule per s.19
In another study Mishra et al.27 have reported rate coefficients for H-atom abstraction from the ethyl group in CF2ClC(O)OCH2CH3 by Cl atoms at 298 K. They reported rate coefficients of 3.88 × 10−14 and 1.82 × 10−12 cm3 per molecule per s for the reaction channels CF2ClC(O)OCH2CH3 + Cl → CF2ClC(O)OCH2CH2 + HCl and CF2ClC(O)OCH2CH3 + Cl → CF2ClC(O)OCHCH3 + HCl, respectively. Mishra et al.27 reported a value of 1.86 × 10−12 cm3 per molecule per s for the overall rate coefficient of the reaction (i.e. the sum of the rate coefficients for each reaction channel) which is in very good agreement with our previous experimental value of (1.5 ± 0.3) × 10−13 cm3 per molecule per s16 and the rate coefficient of (1.55 ± 0.44) × 10−13 cm3 per molecule per s reported in this work.
The RC(O)O– group is known to have a deactivating effect on C–H bonds positioned both α and also β to the group.28 It is interesting to compare the rate coefficients for Cl with the two chlorodifluoroesters with those of methyl acetate (MA) and ethyl acetate (EA). The reactions of Cl with MA and EA have rate coefficients of 2.2 × 10−12 and 1.76 × 10−11 cm3 per molecule per s, respectively, whereby the contribution from H-atom abstraction from the acetyl group CH3C(O)O is <1 × 10−13.28 The rate coefficients for the reactions of Cl atoms with MCDFA and ECDFA are factors of approximately 22 and 11 slower than those of Cl with MA and EA, respectively, indicating the strong deactivating effect of the ClF2C– group on the reactivity of the chlorodifluoroesters toward Cl. It is also interesting to note when comparing rate coefficients for halogenated esters23 that k(Cl + ClF2CC(O)OCH3) > k(Cl + F3CC(O)OCH3) and k(Cl + ClF2CC(O)OCH2CH3) > k(Cl + F3CC(O)OCH2CH3) supporting an expected stronger negative inductive effect for F3C– over ClF2C–.
Product distribution studies
Fig. 2 shows typical product spectra recorded in the Cl-atom initiated oxidation of CF2ClC(O)OCH3. Trace (A) shows a product spectrum obtained after irradiation of a Cl2/chlorofluoroester/air mixture and subtraction of residual MCDFA bands. It can be seen from a comparison of the spectrum with reference spectra of chlorodifluoroacetic acid (trace (B)) and carbon monoxide (trace (C)) that these two compounds are products formed in the Cl + MCDFA reaction. Trace (D) is the residual spectrum that is obtained after subtraction of known products from the spectrum in trace (A).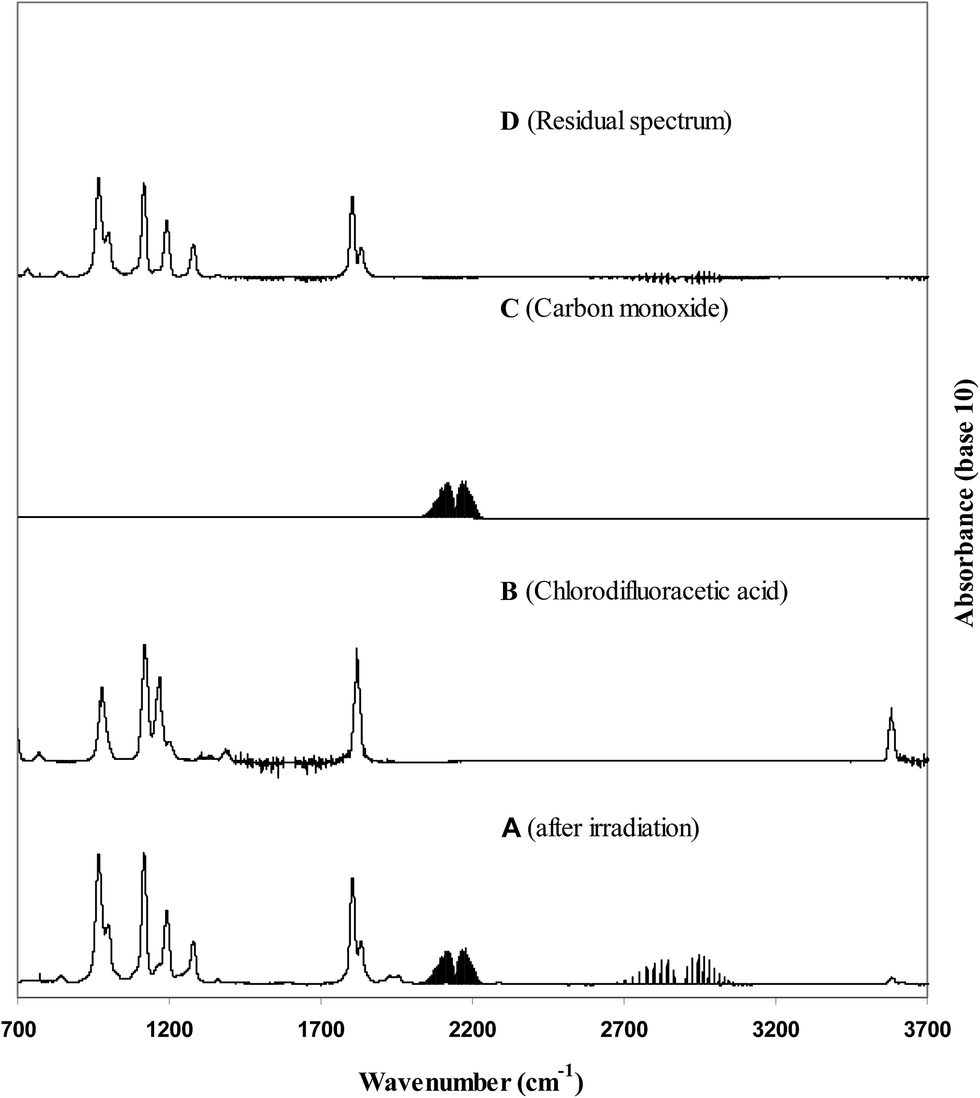 | ||
| Fig. 2 Trace (A) shows the infrared spectrum of a MCDFA/Cl2/air reaction mixture after irradiation and subtraction of residual methyl chlorodifluoroacetate. Trace (B) shows a reference spectra of chlorodifluoroacetic acid (CF2ClC(O)OH), trace (C) a reference spectrum of carbon monoxide and trace (D) the residual product spectrum obtained after subtraction of features due to chlorodifluoroacetic acid and carbon monoxide from the spectrum in trace (A). | ||
Concentration–time profiles of MCDFA and the identified products are shown in Fig. S3.† Plots of the concentration of the identified products as a function of reacted MCDFA (Fig. S4 in ESI†) are linear supporting that they are primary products. From a minimum of 3 experiments averaged molar formation yields of (34 ± 5)% and (26 ± 3)% were obtained for chlorodifluoracetic acid and carbon monoxide, respectively.
In the reaction of Cl atoms with ethyl chlorodifluoroacetate, chlorodifluoracetic acid and carbon monoxide have been also identified as products. Fig. S5,† trace (A), shows an IR spectra acquired after irradiation of a mixture of ECDFA and Cl2 in air where residual absorptions due to ECDFA have been subtracted. Fig. S5,† trace (B), shows a reference spectra of chlorodifluoroacetic acid (CF2ClC(O)OH), trace (C) a reference spectra of carbon monoxide and trace (D) the residual spectrum that is obtained after subtraction of known products from the spectrum in trace (A).
Concentration–time profiles of ECDFA and the identified products chlorodifluoroacetic acid and carbon monoxide are shown in Fig. S6 in the ESI.† The concentration–time contour of chlorodifluoroacetic acid and the linearity of the plot of its concentrations as a function of reacted ECDFA support that it is a primary product. Carbon monoxide shows the same behavior, however, as discussed later the CO is thought to result from the fast reaction of Cl atoms with HCHO. Analysis of the slopes of the plots shown in Fig. S7† give product yields of (86 ± 8)% for chlorodifluoroacetic acid and (34 ± 6)% for carbon monoxide in the reaction of Cl with ethyl chlorodifluoroacetate.
Table 2 lists the yields of the products formed in the reactions of MCDFA and ECDFA with Cl atoms. The errors quoted are a combination of the 2σ statistical errors from the regression analysis plus a contribution to take into account the uncertainty in the spectral subtraction procedure.
| Chlorodifluoroacetate | Products identified | Yield (%) |
|---|---|---|
| CF2ClC(O)OCH3 (methyl chlorodifluoroacetate) | CF2ClC(O)OH | 34 ± 5 |
| CO | 26 ± 3 | |
| CF2ClC(O)OCH2CH3 (ethyl chlorodifluoroacetate) | CF2ClC(O)OH | 86 ± 8 |
| CO | 34 ± 6 |
In the atmosphere the oxidation of the saturated esters is mainly initiated by reaction with OH radicals and proceeds via H-atom abstraction from the alkyl groups (–CH–, –CH2–, –CH3) in the ester forming alkyl radicals. The reactions of the haloesters studied with Cl atoms are less selective than the analogous OH reactions but the subsequent chemistry is similar. Thus, although reactions of Cl-atoms with the esters are not so atmospherically important as the OH reactions, studies of their mechanisms are still necessary and are a convenient way to emulate OH-radical initiated chemistry.
Under atmospheric conditions alkyl radicals react with oxygen to give peroxy radicals. The peroxy radicals formed can react with NO, NO2, HO2, and other peroxy radicals forming, to a large extent, the corresponding alkoxy radicals.29 In the present NOx free reaction systems the alkoxy radicals will be formed by reaction of the peroxy radicals with RO2/HO2 and those shown in Schemes 2 and 3 for MCDFA and ECDFA, respectively, are expected to be dominant.
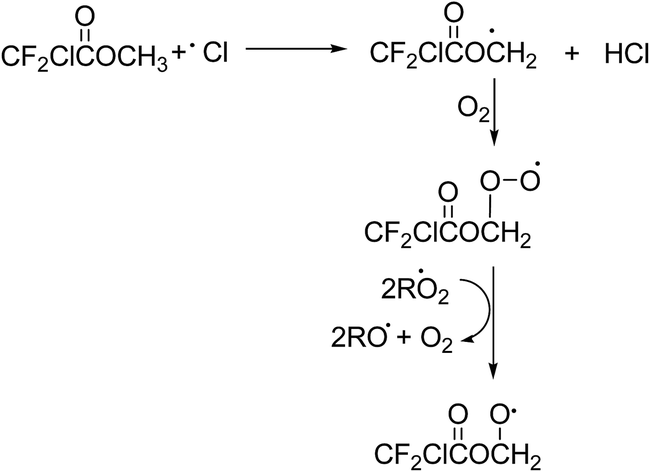 | ||
| Scheme 2 Formation of alkoxy radicals in reaction of Cl + MCDFA. | ||
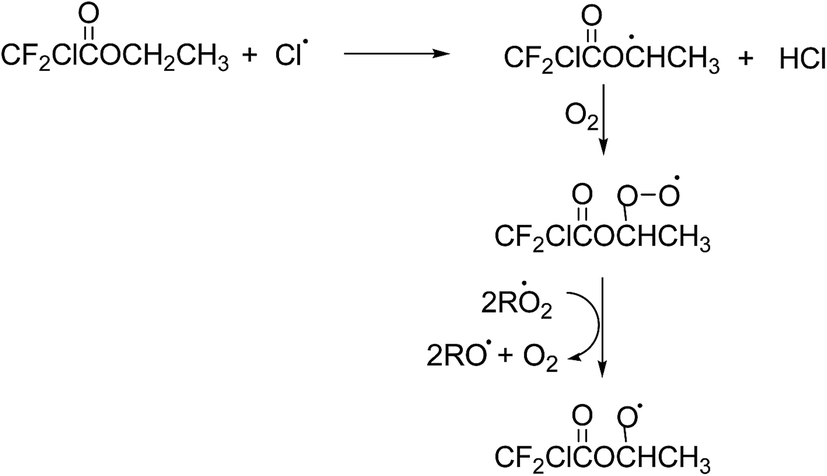 | ||
| Scheme 3 Formation of alkoxy radicals in the reaction of Cl + ECDFA. | ||
Alkoxy radicals are known to react by three different pathways: (i) reaction with oxygen forming a carbonyl compound and HO2 radicals, (ii) decomposition via C–C bond cleavage producing a stable product and a radical and, (iii) if possible, isomerization via a six-membered transition state producing polyfunctional compounds. In addition in the case of esters, the alkoxy radical, formed by H-atom abstraction from the carbon attached to the non-carbonyl oxygen of the ester entity, can undergo what is known as an α-ester rearrangement30 to form an acid and a radical. The products identified in the irradiation of MCDFA/Cl2/air and ECDFA/Cl2/air mixtures allow the elucidation of the fate of the alkoxy radicals.
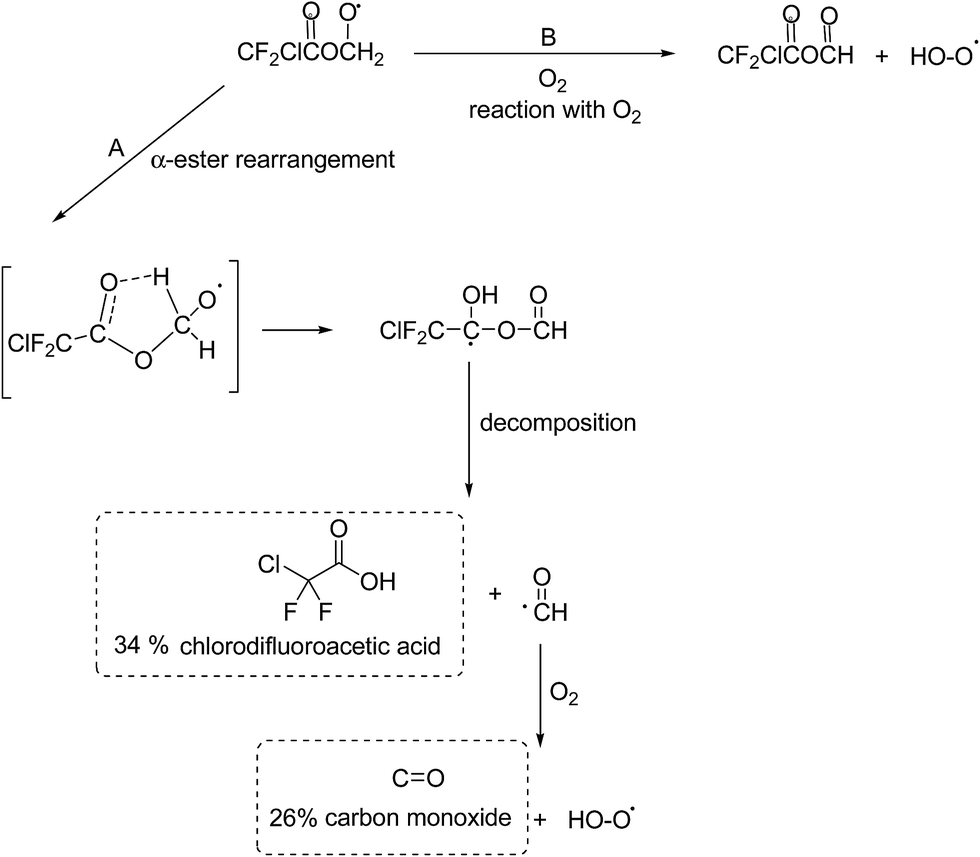 | ||
| Fig. 3 Atmospheric fate of the alkoxy radicals formed after H-atom abstraction by Cl from the –CH3 group in MCDFA. The dashed boxes indicate the identified and quantified products formed. | ||
The possible C–O bond cleavage decomposition channel of the CF2ClC(O)OCH2O˙ radical to form the CF2ClC(O)O˙ radical and HCHO has been omitted in Fig. 3 since formation of formaldehyde was not observed in the reaction system. The CF2ClC(O)O˙ radical can decompose to form CO2 and the CF2Cl˙ radical which can react with O2 to form carbonyl fluoride (CF2O).32 Carbonyl fluoride was observed in the system but its formation was clearly from secondary reactions. In addition, a previous study on the fate of the analogous alkoxy radical CH3C(O)OCH2O˙ formed in the oxidation of methyl acetate has shown that the decomposition channel was also negligible.31
The reaction of CF2ClC(O)OCH2O˙ radicals with O2 will lead to the formation of the mixed acid anhydride CF2ClC(O)OC(O)H (Fig. 3, channel B). Since a reference spectrum for CF2ClC(O)OC(O)H is not commercially available, it was not possible to directly assign absorptions bands in the product spectrum to this compound. Preparative methods are known for mixed anhydrides such as CF2ClC(O)OC(O)H33 unfortunately infrared spectra for the compounds have not yet been reported and it was beyond the scope of the present work to synthesize CF2ClC(O)OC(O)H. However, anhydrides are known important products in the Cl-atom mediated oxidation of alkyl acetates and alkyl haloacetates.30,31 The residual product spectrum bares many similarities with that obtained in the study of Cl atoms with CF3C(O)OCH3 where the main absorptions bands were assigned to formic trifluoroacetic anhydride CF3C(O)OCHO.32
The unique structural feature of an anhydride is the presence of two carbonyl groups linked to each other via an oxygen atom. This structure gives rise to two carbonyl absorptions in the infrared arising from symmetrical and asymmetrical stretching of the carbonyl groups. The residual product spectrum from Cl + MCDFA contains two carbonyl absorptions at 1831 and 1805 cm−1 separated by 26 cm−1 whereby the absorption at 1805 cm−1 is stronger than that at 1831 cm−1. The small carbonyl band splitting is expected for anhydrides of structure RC(O)OCHO.34 In the fingerprint region of the product spectrum band maxima occur at ∼1279, ∼1193, ∼1115, ∼999, ∼966 cm−1 whereby those at 1115 and 966 cm−1 are the strongest and equally intense. Interpretation of anhydride spectra in the fingerprint region is complicated because of a large number of C–O and C–C stretchings and various bending modes, many of which are strongly mixed. The strong band at 1115 cm−1 is an indication for the presence of an anhydride and is generally associated with C–O and C–C vibrations. However, although many anhydrides have strong bands in this region it is not a unique marker for the presence of an anhydride since many other compounds absorb in this region.35,36 The bands at 1279 and 1193 cm−1 are consistent with vibrations from C–F and C–Cl in the ClF2C– group: similar bands occur in the infrared spectra of formic trifluoroacetic anhydride and trifluoroacetic anhydride.35–37 The absorption bands at ∼999 and ∼960 cm−1 are most probably also from C–F and C–Cl vibrations, halogenated compounds such as 1,2-dichloro-tetrafluoroethane show fairly strong bands in this region.
An infrared spectrum has been simulated for CF2ClC(O)OC(O)H using the using Gaussian 03 ab initio quantum chemistry package.38 The spectrum was calculated using the B3LYP/6 31+G level of theory.39–41 The simulated infrared spectrum is compared in Fig. S8,† panel B, with the residual product spectrum (panel A) from the Cl + MCDFA reaction. Although the computed spectrum captures the main spectral features of the experimental absorption bands reasonably well, there are large discrepancies in the absorption frequencies especially for the carbonyl bands. The computed spectrum, however, does lend additional support to the other spectral experimental evidence that the residual product spectrum is very probably mainly due to formation of CF2ClC(O)OC(O)H from reaction of CF2ClC(O)OCH2O˙ radicals with O2. Secondary reactions of CF2ClC(O)OC(O)H with Cl are the most probable source of the small amounts of F2CO observed in the reaction system.
The experimental evidence in this work, i.e. the high formation yield of chlorodifluoroacetic acid, also supports that in the reaction of Cl with ECDFA attack of the Cl atom at the –CH2– group dominates. The CF2ClC(O)OCH·CH3 radical thus formed will add O2 to form a peroxy radical further reactions of which with RO2/HO2 will yield to a large extent the alkoxy radical CF2ClC(O)OCHO·CH3. A simplified mechanism showing the further possible reaction pathways of the CF2ClC(O)OCHO·CH3 radical is presented in Fig. 5.
The observation of a high chlorodifluoroacetic acid yield (∼85%) supports that the α-ester rearrangement (Fig. 4, channel A) is the main reaction channel for the CF2ClC(O)OCHO·CH3 radical, assuming that no other source of the acid exists. Blanco et al.32 in their study on the Cl-atom oxidation of fluoroacetates report a value of 78 ± 5% for the formation of trifluoroacetic acid in the reaction of Cl with ethyl trifluoroacetate. Orlando and Tyndall42 have reported a yield of 53% for the formation of acetic acid in the reaction of Cl with ethyl acetate and concluded from their system analysis that other routes to the formation of the acid were very minor. In addition, Tuazón et al.30 and Picquet-Varrault et al.43 have reported yields of 96% and 75% for the formation of acetic acid in the reaction of OH radicals with CH3C(O)OCH2CH3, respectively. All of these studies are consistent with the main reaction pathway being abstraction of an H-atom from the –CH2– group with subsequent chemistry via an α-ester rearrangement leading to the corresponding acid compound.
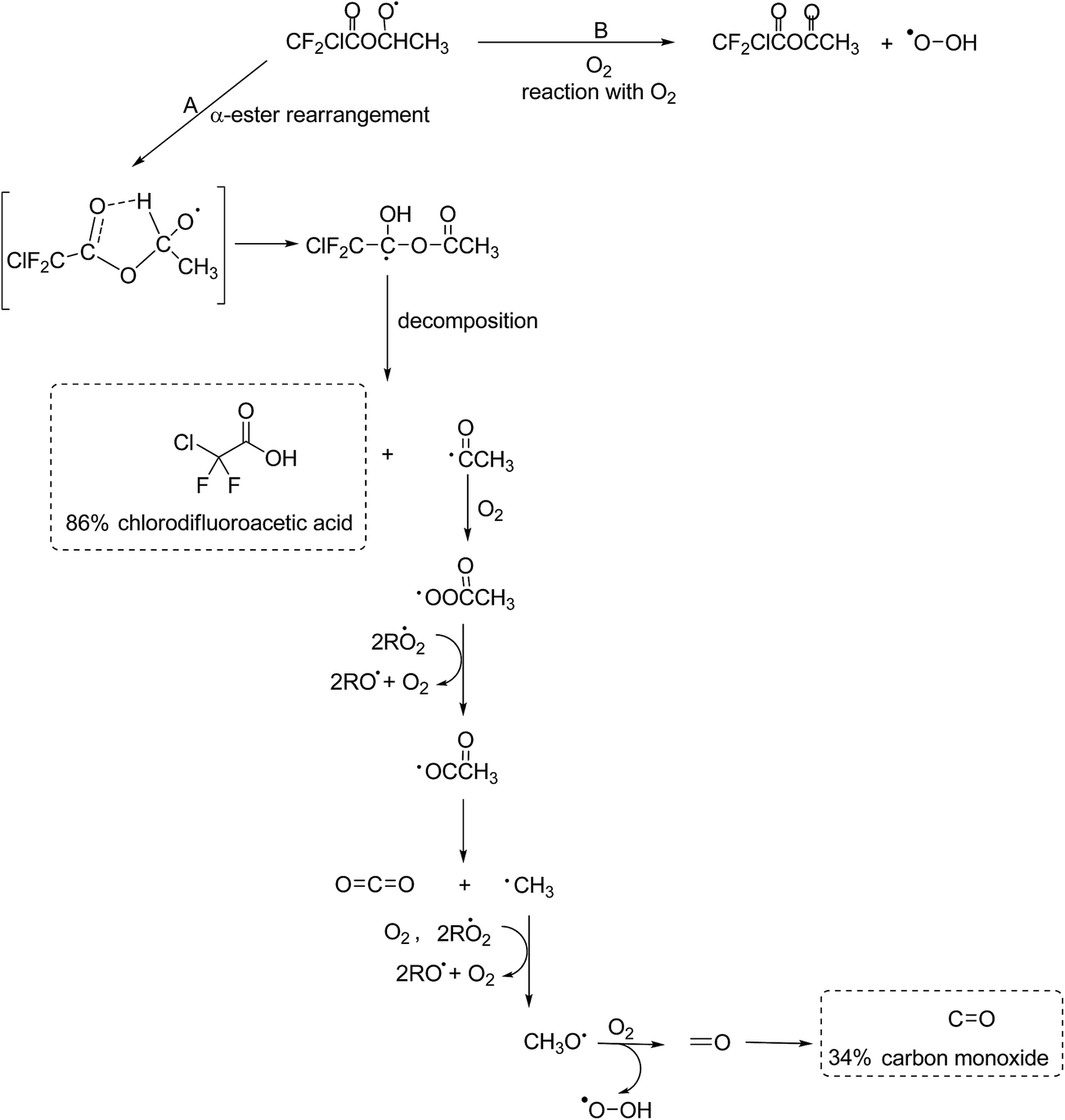 | ||
| Fig. 4 Atmospheric fate of the alkoxy radicals formed after H-atom abstraction by Cl from the –CH2– group in ECDFA. The dashed boxes indicate the identified and quantified products formed. | ||
The co-product of CF2ClC(O)OH, the CH3C(O) radical, will add O2 to form the CH3C(O)OO peroxy radical. This radical can react with HO2 to form peroxy acetic acid (CH3C(O)OOH), or react with other peroxy radicals to form CO2 and CH3 radicals. No evidence for formation of peroxy acetic acid was been observed in the residual product spectrum, however, an OH absorption was observed at ∼3585 cm−1 suggesting formation of a product containing either an OH or OOH group. Although CO2 cannot be quantitatively determined in the system because of high background levels, there where indications that its concentration increased during the course of the reaction. The CH3 radical will be oxidized to formaldehyde (HCHO) the further reaction of which will give CO. Formaldehyde could not be detected in the reaction system, which is not surprising given the high reactivity of HCHO with Cl. Orlando and Tyndall42 in their study on the reaction of Cl with ethyl acetate could only detect traces of HCHO. Interestingly, the acid to CO ratio in this study on Cl with CF2ClC(O)OCH2CH3 and in the studies on Cl with CF3C(O)OCH2CH332 and Cl with CH3C(O)OCH2CH342 are all ∼0.4 indicating other fates for the CH3C(O) radical rather than oxidation through HCHO to CO. It was not possible to elucidate the other reaction pathways from the present FTIR data.
The CF2ClC(O)OCHO·CH3 alkoxy radical could potentially decompose via a C–O–C bond cleavage forming CF2ClC(O)O˙ radicals and acetaldehyde. The CF2ClC(O)O˙ radicals would be expected to decompose to form CO2 and ˙CF2Cl radicals and the subsequent reactions of which would be expected to produce CF2O. Neither the formation of CF2O nor formation of the decomposition co-product acetaldehyde was observed in the product spectra suggesting that the contribution from this channel was negligible. The absence of CF2O also shows that no other reaction product was oxidized through to CF2O in the reaction system.
The absorption bands in the residual product spectrum from the reaction of Cl with ECDFA are not as distinct as in the residual product spectrum for Cl with MCDFA with more overlapping of bands which suggests more than one compound is present. Indeed, a comparison of the residual product spectrum from Cl with MCDFA, which has been identified as belonging mainly to the anhydride ClCF2C(O)OCHO, with that of Cl with ECDFA shows that ClCF2C(O)OCHO is formed in the reaction of Cl with ECDFA. After subtraction of the absorptions due to ClCF2C(O)OCHO, the following absorptions remain at ∼1864, ∼1819, ∼1302, ∼1169, ∼1125, ∼1093 (shoulder) and ∼978 cm−1. The two carbonyl absorptions at 1864 and 819 cm−1 are separated by 45 cm−1, whereby 1819 cm−1 is the stronger of the two. As discussed for the assignment of CClF2C(O)OC(O)H as a product in the reaction of Cl with MCDFA, bands at ∼1302, ∼1169 and ∼978 cm−1 are consistent with C–F and C–Cl vibrations from the ClF2C– group and a strong absorption at ∼1125 cm−1 would fit with a C–O–C type vibration. These observed absorptions are consistent with the formation of the anhydride CF2ClC(O)OC(O)CH3 from the reaction of CF2ClC(O)OCHO·CH3 radical with O2 (Fig. 4, channel B). This anhydride is not commercially available for quantification purposes, however, the high yield of CF2ClC(O)OH and evidence for the formation of CF2ClC(O)OC(O)H suggest that its yield is probably <10%.
Atmospheric implications
Methyl chlorodifluoracetate and ethyl chlorodifluoroacetate are removed from the troposphere mainly by their rapid reactions with OH radicals16 which results in atmospheric lifetimes of the order of 10–50 days. However, in some marine and continental regions close to industrial emissions, peak values of 1 × 105 atoms per cm3 for Cl atoms concentrations have been observed.3–11 Under such circumstances the reaction of Cl with the chlorofluoroacetates can be a significant if not dominant loss process for the compounds. Photolysis is not an important atmospheric loss process for these chlorofluorinated compounds since they do not absorb radiation at actinic wavelengths.16As reported for other haloesters44,45 we expect relatively low values of the Henry laws constants at 298 K for both compounds, indicating that wet deposition of these chlorofluoro acetates will not be significant compared to the OH-radical or Cl-atom initiated degradation. The volatility of these compounds will render dry deposition an unlikely removal mechanism.
The temperature dependences of the Cl-rate coefficients reported in this work were used to calculate the rate of loss of the chlorofluoroacetates studied by their tropospheric reaction with Cl atoms. At a given temperature (T) corresponding to a given altitude in the troposphere, the rate of loss of the chlorofluoracetates is defined as the product between the Cl rate coefficient (T) and the [Cl] at this altitude.
Using a 24 h daytime average global tropospheric Cl atom concentration of [Cl] = 1 × 104 atoms per cm3,46 the Arrhenius parameters reported in this work and considering a lapse rate in the troposphere of −6.5 K km−1,47 we have calculated the temperature profile between 0 and 10 km. Table S1† shows the Cl removal rates for the chlorofluoroacetates studied as a function of altitude in the troposphere considering temperature of 298.15 K at 0 km. The results showed in Table S1† are plotted in the Fig. 5.
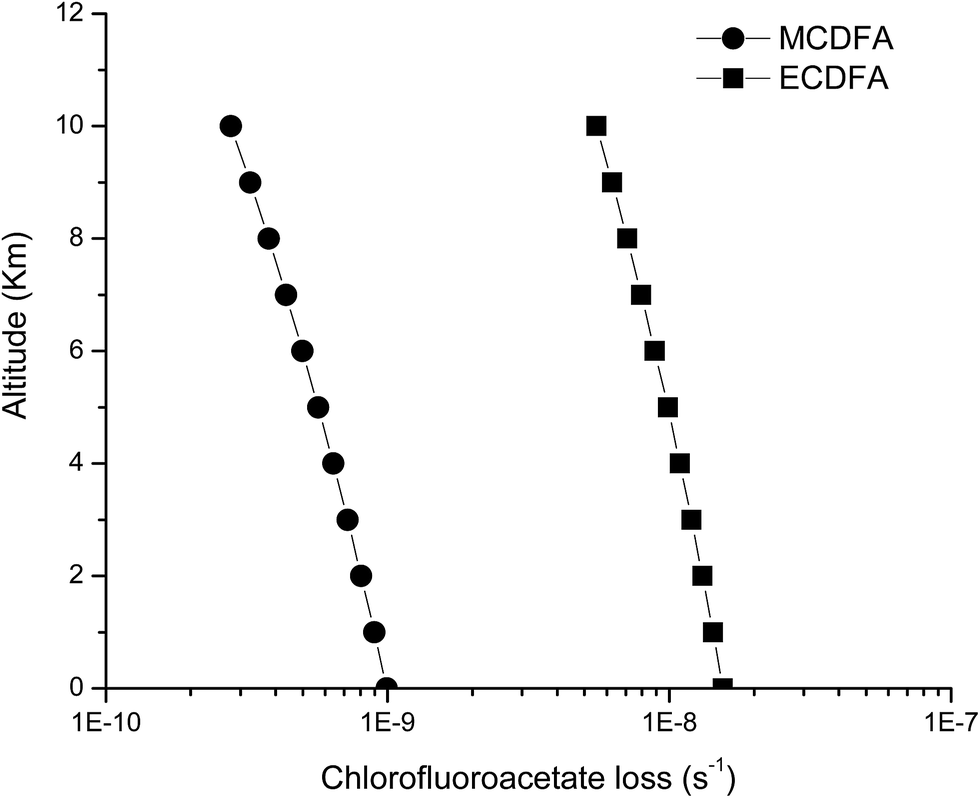 | ||
| Fig. 5 Loss rates for methyl chlorodifluoracetate (●) and ethyl chlorodifluoracetate (■) through reaction with Cl atoms as a function of altitude (km) of the atmosphere. | ||
The loss rates (in s−1) of the haloacetates studied at sea level (0 km) are 9.93 × 10−10 and 1.55 × 10−8 and near to the tropopause (∼10 km) around 2.78 × 10−10 and 5.51 × 10−9 for methyl chlorodifluoroacetate and ethylchlorodifluoroacetate, respectively.
The positive temperature dependence of the reactions of Cl atoms with both MCDFA and ECDFA is consistent with the dominance of a hydrogen atom abstraction mechanism.
The product investigations presented in this work have shown that the major product of the Cl-atom initiated oxidation of methyl chlorodifluoroacetate under NOx-free conditions will be formic dichlorofluoroacetic anhydride (CF2ClC(O)OC(O)H). We expect that chlorofluoro anhydride will be further oxidized fairly rapidly by reaction with OH radicals and may also photolyze since the compound contains the –C(O)H chromophore. The other product observed in the reaction of Cl with MCDFA was chlorodifluoracetic acid (∼30%). Both, anhydrides and acids are highly soluble compounds and may be rapidly incorporated into cloud droplets (where hydration of the anhydrides leads to acids) and rain out, contributing to the precipitation acidity.
For the reaction of Cl with ETFA the main product observed was chlorodifluoroacetic acid (∼88%). Haloacetic acids are ubiquitous environmental pollutants and their presence in the environment is due both to natural processes and to human activities. They partition into water because of their large Henry's law constants.48,49 They could be accumulated to relatively high concentrations in aqueous reservoirs and would likely accumulate to toxic levels. Furthermore, haloacetic acids have been detected in many places such as soil, conifer needles and lichens, snow and ice in Antarctica, fog and rainwater, sea water, lake water, wastewater and drinking water.50,51 Haloacetic acids, as chlorodifluoracetic acid are generally phytotoxic and toxic to green algae,52 while some haloacetic acids, e.g., dichloroacetic acid and trichloracetic acid, have hepatocarcinogenic potential.53
In this sense, studies of the tropospheric degradation of chlorofluoroacetates are important and necessary, since these compounds break down relatively rapidly to several products including persistent substances such as chlorodifluoroacetic acid. Concentrations of CDFA with a maximum of 0.0002 ng L−1 were reported in water samples taken in Canada.48 However, a continuing monitoring of the environmental concentrations of CDFA is recommended to confirm trends in surface and rainwater.
In addition, further studies on the product distributions from the OH-radical and Cl-atom initiated oxidation of haloesters need to be performed over the range of temperatures and NOx levels typically prevailing in the troposphere in order to gain a better quantitative understanding of their contribution to photooxidant formation and precipitation acidity.
Acknowledgements
The authors wish to acknowledge Deutsche Forschungsgemeinschaft (DFG), DAAD-MINCyT, FONCyT and CONICET for financial support of this research. M. B. Blanco wishes to acknowledge to the Alexander von Humboldt Foundation for support through a postdoctoral fellowship.References
- W. T. Tsai, Environmental risk assessment of hydrofluoroethers (HFEs), J. Hazard. Mater., 2005, 119, 69–78 CrossRef CAS PubMed.
- A. Sekiya and S. Misaki, J. Fluorine Chem., 2000, 101, 215–221 CrossRef CAS.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS.
- W. C. Keene, A. A. P. Pszenny, D. J. Jacob, R. A. Duce, J. N. Galloway, J. J. Schultz Tokos, H. Sievering and J. F. Boatman, Global Biogeochemistry Cycles, 1990, vol. 4, pp. 407–430 Search PubMed.
- B. J. Finlayson-Pitts, Res. Chem. Intermed., 1993, 19, 235–249 CrossRef CAS.
- C. George, W. Behnke and C. Zetzsch, ChemPhysChem, 2010, 11, 3059–3062 CrossRef CAS PubMed.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464, 271–274 CrossRef CAS PubMed.
- H. D. Osthoff, J. M. Roberts, A. R. Ravishankara, E. J. Williams, B. M. Lerner, R. Sommariva, T. S. Bates, D. Coffman, P. K. Quinn, J. E. Dibb, H. Stark, J. B. Burkholder, R. K. Talukdar, J. Meagher, F. C. Fehsenfeld and S. S. Brown, Nature Geoscience, 2008, 1, 324–328 CrossRef CAS.
- G. J. Phillips, M. J. Tang, J. Thieser, B. Brickwedde, G. Schuster, B. Bohn, J. Lelieveld and J. N. Crowley, Geophys. Res. Lett., 2012, 39, L10811, DOI:10.1029/2012gl051912.
- C. J. Young, R. A. Washenfelder, J. M. Roberts, L. H. Mielke, H. D. Osthoff, C. Tsai, O. Pikelnaya, J. Stutz, P. R. Veres, A. K. Cochran, T. C. VandenBoer, J. Flynn, N. Grossberg, C. L. Haman, B. Lefer, H. Stark, M. Graus, J. de Gouw, J. B. Gilman, W. C. Kuster and S. S. Brown, Environ. Sci. Technol., 2012, 46, 10965–10973 CrossRef CAS PubMed.
- E. Galán, I. González and B. Fabbri, Estimation of fluorine and chlorine emissions from Spanish structural ceramic industries, Atmos. Environ., 2002, 36, 5289–5298 CrossRef.
- G. James, M. Neil Jr and D. J. Burton, J. Fluorine Chem., 1991, 55, 225–227 CrossRef.
- D. Desbouis, R. Hardré and M. Réglier, Ethyl chlorodifluoroacetate. e-EROS Encyclopedia of Reagents for Organic Synthesis, 2009, DOI:10.1002/047084289X.rn00987.
- N. Oyaro, S. R. Sellevag and C. J. Nielsen, Environ. Sci. Technol., 2004, 38, 5567–5576 CrossRef CAS PubMed.
- M. P. Sulback Andersen, O. J. Nielsen, T. J. Wallington, M. D. Hurley and G. W. DeMoore, J. Phys. Chem. A, 2005, 109, 3926–3934 CrossRef PubMed.
- M. B. Blanco and M. A. Teruel, Chem. Phys. Lett., 2007, 441, 1–6 CrossRef CAS.
- J. W. Martin, J. Franklin, M. L. Hanson, K. R. Solomon, S. A. Mabury, D. A. Ellis, B. F. Scott and D. C. G. Muir, Environ. Sci. Technol., 2000, 34, 274–281 CrossRef CAS.
- I. Barnes and K. H. Becker, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Jr Hampson, J. A. Kerr, M. J. Rossi and J. Troe, IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry Web Version, Dec 2001 Search PubMed.
- A. Aranda, Y. Díaz de Mera, I. Bravo, D. Rodríguez, A. Rodríguez and E. Martínez, Environ. Sci. Technol., 2006, 40, 5971–5976 CrossRef CAS PubMed.
- Y. Díaz de Mera, A. Aranda, I. Bravo, E. Moreno, E. Martínez and A. Rodríguez, Chem. Phys. Lett., 2009, 479, 20–24 CrossRef.
- A. Rodríguez, D. Rodríguez, A. Moraleda, I. Bravo, E. Moreno and A. Notario, Atmos. Environ., 2014, 96, 145–153 CrossRef.
- M. B. Blanco, I. Barnes, P. Wiesen and M. A. Teruel, Chem. Phys. Lett., 2015, 638, 15–20 CrossRef CAS.
- B. K. Mishra, A. K. Chakrabartty and R. C. Deka, J. Mol. Model., 2013, 19, 3263–3270 CrossRef CAS PubMed.
- E. P. Z. Wigner, Phys. Chem., 1932, B19, 203–216 CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- B. K. Mishra, A. K. Chakrabartty and R. C. Deka, Struct. Chem., 2014, 25, 463–470 CrossRef CAS.
- J.-H. Xing, K. Takahashi, M. D. Hurley and T. J. Wallington, Chem. Phys. Lett., 2009, 474, 268–272 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- E. C. Tuazón, S. M. Aschmann, R. Atkinson and W. P. L. Carter, J. Phys. Chem., 1998, 102, 2316–2321 CrossRef.
- L. K. Christensen, J. C. Ball and T. J. Wallington, J. Phys. Chem. A, 2000, 104, 345–351 CrossRef CAS.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Atmospheric photooxidation of fluoroacetates as a source of fluorocarboxylic acids, Environ. Sci. Technol., 2010, 44, 2354–2359 CrossRef CAS PubMed.
- J. Codnia and M. L. Azcárate, Photochem. Photobiol., 2006, 82, 755–762 CrossRef CAS PubMed.
- S. Montiel-Smith, S. Meza-Reyes, O. Viňas-Bravo, M. A. Fernández-Herrera, R. Martinez-Pascual, J. Sandoval-Ramírez, A. Fuente, M. Reyes and J. A. Ruiz, ARKIVOC, 2005, vi, 127–135 Search PubMed.
- G. Wu, S. Shlykov, C. Van Alsenoy, H. J. Geise, E. Sluyts and B. J. Van der Veken, J. Phys. Chem., 1996, 100, 11620–11629 CrossRef CAS.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Chapman and Hall, London, II, 2nd edn, 1980 Search PubMed.
- R. L. Redington and K. C. Lin, Spectrochim. Acta, Part A, 1971, 27, 2445–2460 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Jr Vreven, N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. TomasiV. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli; J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian Inc., Wallingford, CT, 2004.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Int. J. Chem. Kinet., 2010, 32, 397–413 CrossRef.
- B. Picquet-Varrault, J. F. Doussin, R. Durand-Jolibois and P. Carlier, Phys. Chem. Chem. Phys., 2001, 3, 2595–2606 RSC.
- S. Kutsuna, L. Chen, K. Ohno, K. Tokuhashi and A. Sekiya, Atmos. Environ., 2004, 38, 725–738 CrossRef CAS.
- S. Kutsuna, L. Chen, T. Abe, J. Mizukado, T. Uchimaru, K. Tokuhashi and A. Sekiya, Atmos. Environ., 2005, 39, 5884–5892 CrossRef CAS.
- O. W. Wingenter, M. K. Kubo, N. J. Blake, T. W. Smith, D. R. Blake and F. S. Rowland, J. Geophys. Res., 1996, 101, 4331–4340 CrossRef CAS.
- M. Beychok, 2013, http://www.eoearth.org/view/article/170859.
- J. W. Martin, J. Franklin, M. L. Hanson, K. R. Solomon, S. A. Mabury, D. A. Ellis, B. F. Scott and D. C. G. Muir, Environ. Sci. Technol., 2000, 34, 274–281 CrossRef CAS.
- D. J. Bowden, S. L. Clegg and P. Brimblecombe, J. Atmos. Chem., 1998, 29, 85–107 CrossRef CAS.
- R. J. B. Peters, J. Environ. Monit., 2003, 5, 275–280 RSC.
- S. Hashimoto, T. Azuma and A. Otsuki, Environ. Toxicol. Chem., 1998, 17, 798–805 CrossRef CAS.
- M. L. Hanson and K. R. Solomon, Environ. Pollut., 2004, 130, 371–383 CrossRef CAS PubMed.
- S. L. Herren-Freund, M. A. Pereira, M. D. Khoury and G. Olson, Toxicol. Appl. Pharmacol., 1987, 90, 183–189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03454c |
| This journal is © The Royal Society of Chemistry 2016 |