
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Delivery of siRNA targeting HIF-1α loaded chitosan modified D-α-tocopheryl polyethylene glycol 1000 succinate-b-poly(ε-caprolactone-ran-glycolide) nanoparticles into nasopharyngeal carcinoma cell to improve the therapeutic efficacy of cisplatin
Daizheng
Lian†
a,
Yuhan
Chen†
b,
Gang
Xu†
a,
Xiaowei
Zeng
c,
Zhuangling
Li
a,
Zihuang
Li
a,
Yayan
Zhou
a,
Lin
Mei
c and
Xianming
Li
*a
aDepartment of Radiation Oncology, Second Clinical Medicine College of Jinan University, Shenzhen, Guangdong 518020, PR China. E-mail: lixianming1828@hotmail.com; Fax: +86 75525533018; Tel: +86 75525533018
bDepartment of Radiation Oncology, Zhongshan Hospital, Fudan University, Shanghai 200032, PR China
cThe Shenzhen Key Lab of Gene and Antibody Therapy, Division of Life and Health Sciences, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, PR China
First published on 5th April 2016
Abstract
Hypoxia inducible factor-1α (HIF-1α) related signaling pathways mediating chemoresistance has been found in various kinds of cancer, including nasopharyngeal carcinoma (NPC). In this research, a chitosan modified D-α-tocopheryl polyethylene glycol 1000 succinate-b-poly(ε-caprolactone-ran-glycolide) (TPGS-b-(PCL-ran-PGA)) nanoparticle (NP) was prepared for small interfering ribonucleic acid (siRNA) targeting HIF-1α delivery. The results showed that chitosan-modified TPGS-b-(PCL-ran-PGA) NPs could effectively deliver siRNA into CNE-2 cells, resulting in the decrease of HIF-1α expression and cell viability. Decreased sensitivity of cisplatin in CNE-2 cells under hypoxia condition was correlated with the overexpression of HIF-1α and multiple drug resistance gene 1 (MDR1)/P-glycoprotein (P-gp). Inhibiting HIF-1α by siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs significantly decreased the expression of HIF-1α and MDR1/P-gp and restored the effect of cisplatin on CNE-2 cells. Moreover, synergistic anti-tumor effects could be achieved by the combined use of siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs and cisplatin. These findings showed that the chitosan modified TPGS-b-(PCL-ran-PGA) NPs could function as an effective carrier for siRNA delivery aiming at modulating HIF-1α expression to increase the therapeutic potential of cisplatin in NPC therapy.
1. Introduction
Nasopharyngeal carcinoma (NPC) is one of the most common head and neck cancers in China and south Asia. Radiotherapy in combination with cisplatin-based chemotherapy is the dominant treatment for advanced NPC.1 Local recurrence and distant metastasis still remain the major reason for the poor survival of advanced NPC patients, indicating that there exists resistance of NPC cells toward radiotherapy and chemotherapy.2 It has been proven that hypoxia is a common biological feature of many malignancies and hypoxia microenvironment influences many aspects of tumor biology, including energy metabolism, angiogenesis, invasiveness and metastasis, resulting in hypoxia induced radio- and chemoresistance in cancer therapy.3 Hypoxia inducible factor-1α (HIF-1α) is a crucial transcription factor mediating the adaptive response to hypoxia condition and overexpression of HIF-1α was correlated with the poor survival of many cancers, including NPC.4,5 Previous researches had revealed that chemoresistance could be directly or indirectly induced by HIF-1α mediated vascular endothelial growth factor (VEGF), glucose transporter 1 (Glut-1), multiple drug resistance gene 1 (MDR1), and B-cell lymphoma 2 (Bcl-2) pathway.6 HIF-1α induced cisplatin resistance was found in many kinds of tumor, such as neuroblastoma, osteosarcoma, laryngeal cancer, gastric cancer, non-small cell lung cancer and NPC.7–12 Thus, HIF-1α becomes a candidate target to reverse hypoxia-induced cisplatin resistance.Using small interfering ribonucleic acid (siRNA) to silence overexpressed proto-oncogenes or oncogenes in solid tumor cells is popular in cancer treatment experiments.13 Various kinds of nanoparticles (NPs) are considered to be biodegradable, biocompatible, control released and low toxicity, which are now widely adopted as siRNA delivery system.14–16 A number of studies have reported that with the surface modification of NPs, such as cationic modification, could form effective nanosize delivery systems with increasing encapsulation of gene materials and promote the electrostatic interaction with the negatively charged cell surface contributing to improve the cellular uptake.17 Chitosan is commonly approached as the cationic polymer because of its biological adhesive properties and ability to enhance the stability of macromolecules as well as to control the release of drugs, proteins, and peptides.18–20 Emerging evidences have confirmed that chitosan modified NPs delivery systems could facilitate anti-cancer agents to exert more excellent therapeutic efficiency than the unmodified NPs.21,22
In our previous research, D-α-tocopheryl polyethylene glycol 1000 succinate-b-poly(ε-caprolactone-ran-glycolide) (TPGS-b-(PCL-ran-PGA)) NP was successfully synthesized as a delivery system of siRNA targeting HIF-1α for NPC gene therapy and the siRNA loaded NPs significantly decreased the expression level of HIF-1α and efficiently suppressed tumor growth.23 In the present study, we delivered siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs into human NPC cell line CNE-2 to evaluate the in vitro therapeutic effects of HIF-1α inhibition in combination with cisplatin for NPC.
2. Materials and methods
2.1 Materials
TPGS-b-(PCL-ran-PGA) copolymer (Mw approximately 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was synthesized in our laboratory (Tsinghua University, China). TPGS, glycolide (1,4-dioxane-2,5-dione), poly(vinyl alcohol) (PVA; 80% hydrolyzed), chitosan (molecular weight of 140–220 kDa) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St. Louis, MO, USA). ε-CL was purchased from Acros Organics (Geel, Belgium). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was obtained from VECTOR (Burlingame, CA, USA). RPMI medium 1640 basic, fetal bovine serum (FBS), penicillin–streptomycin, and phosphate-buffered saline (PBS) were procured from Invitrogen-Gibco (Carlsbad, CA, USA). All other chemicals used in the study were of analytical grade.
000) was synthesized in our laboratory (Tsinghua University, China). TPGS, glycolide (1,4-dioxane-2,5-dione), poly(vinyl alcohol) (PVA; 80% hydrolyzed), chitosan (molecular weight of 140–220 kDa) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St. Louis, MO, USA). ε-CL was purchased from Acros Organics (Geel, Belgium). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was obtained from VECTOR (Burlingame, CA, USA). RPMI medium 1640 basic, fetal bovine serum (FBS), penicillin–streptomycin, and phosphate-buffered saline (PBS) were procured from Invitrogen-Gibco (Carlsbad, CA, USA). All other chemicals used in the study were of analytical grade.
Human siRNA targeting HIF-1α (sense 5′-GGAAAUGAGAGAAAUGCUUTT-3′, antisense 5′-AAGCAUUUCUCUCAUUUCCTT-3′), scrambled control for HIF-1α siRNA and fluorescence FAM labeled siRNA (FAM-siRNA) were obtained from GenePharma Co., Ltd. (Suzhou, Jiangsu, China). RNase-free diethyl pyrocarbonate (DEPC)-treated Milli-Q water was used for all dilutions.
2.2 Cell culture
CNE-2 cells (ATCC 0384, VA, USA) were cultured in RPMI 1640 (pH 7.4) supplemented with 10 μg mL−1 streptomycin sulfate, 100 μg mL−1 penicillin G, and 10% (v/v) FBS. The cells were incubated at 37 °C in a 5% CO2 humidified atmosphere. For hypoxia induction, cells were incubated with CoCl2 (100 μmol L−1) for 24 h to simulate the tumor hypoxia microenvironment.2.3 Preparation of the siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs
To prepare the chitosan solutions, different amount of chitosan were dissolved in 10 mL DEPC treated water containing 5% glacial acetic acid under magnetic stirring for 4 hours at 40 °C. 1 mL of dichloromethane containing 10 mg of TPGS-b-(PCL-ran-PGA) NPs was added dropwise to various amount of chitosan solutions under magnetic stirring. After 2 h of stirring, the chitosan modified TPGS-b-(PCL-ran-PGA) NPs (CNPs) were collected by centrifugation at 14000 rpm for 15 min at 4 °C. The siRNA loaded CNPs were prepared by a modified double emulsion method. Briefly, 10 mg CNPs were dissolved in 500 μL of dichloromethane, and 200 μL of 5 μM siRNA solution in DEPC-treated Milli-Q water was added. The mixture was then sonicated for 60 s at a 30 W output to form a water-in-oil (w1/o) emulsion. After adding 2 mL of 2% (w/v) PVA into the emulsion, the mixture was then sonicated for another 60 s to form a water-in-oil-in-water (w1/o/w2) double emulsion. The resulting emulsion was subsequently diluted with 4 mL of 2% (w/v) PVA and stirred for 3 h at room temperature to evaporate dichloromethane. Finally, the NPs were harvested by centrifugation at 14000 rpm for 20 min at 4 °C and washed twice with 6 mL of RNase free water to remove the emulsifiers and unencapsulated siRNA.
2.4 Characterization of CNPs
The mean particle size and poly dispersive index (PDI) of NPs were determined using dynamic light scattering (DLS, Zetasizer Nano ZS90, Malvern Instruments Ltd., Malvern, UK). The particle size and zeta potentials were measured using a laser Doppler anemometry (Zetasizer Nano ZS90). The morphological and surface characteristics were examined by Transmission Electron Microscope (TEM). The siRNA encapsulation efficiency (EE) and loading efficiency (LE) were evaluated as the method previously reported.242.5 In vitro release study
The siRNA loaded NPs or siRNA loaded CNPs was dispersed into 1 mL of Tris–EDTA (TE) buffer at pH 7.4 or 4.0 in RNase-free Eppendorf tubes on a rotary shaker at 100 rpm and 37 °C. At predetermined interval 1, 2, 4, 6, 12, 24, 48, 72 and 96 h, the whole media was collected by centrifugation at 12000 rpm for 20 min and replaced with equal volumes of fresh TE buffer to maintain sink conditions. The amount of siRNA released at each time point was determined at 260 nm with an UV spectrophotometer (Evolution 260, Thermo Fisher Scientific, USA, wavelength accuracy: ±0.8 nm).
2.6 Cellular uptake study
Confocal laser microscopy analysis was used to evaluate the cellular uptake and intracellular distribution of siRNA loaded NPs. The siRNA was labeled with 5-carboxyfluorescein (FAM). CNE-2 cells were seeded in 6-well plate and incubated for 24 h at 37 °C. The naked FAM-siRNA, FAM-siRNA loaded NPs or FAM-siRNA loaded CNPs was added separately to the plate. After incubating for 4 h, the sample wells were carefully washed twice and then fixed with 4% paraformaldehyde for 20 min at room temperature. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) for 5 min at room temperature and rinsed three times with PBS. Images were acquired using a confocal laser microscope (Fluoview FV-1000, Olympus Optical Co., Ltd., Tokyo, Japan). The emission wave length of FAM and DAPI was 488 and 340 nm, respectively.2.7 Real time RT-PCR
Total RNA were isolated from cells according to TRIzol reagent protocol (Invitrogen, USA). cDNA was synthesized using the Prime Script RT reagent Kit (Takara Bio, Japan). Quantitative PCR amplification was conducted in a real-time fluorescent measurement system (ABI7300, USA) using the iTaq™ Universal SYBR® Green Supermix (BIO-RAD, USA) according to the protocol provided. The sense and antisense primers used include the following sequences: HIF-1α, 5′-CAAAACACACAGCGAAGC-3′ and 5′-TCAACCCAGACATATCCACC-3′, MDR1, 5′-CTTGGCAGCAATTAGAAC-3′ and 5′-TCAGCAGGAAAGCAGCAC-3′, β-actin, 5′-AGCGAGCATCCCCCAAAGTT-3′ and 5′-GGGCACGAAGGCTCATCATT-3′. The HIF-1α and MDR1 mRNA levels were normalized by β-actin.2.8 Western blot
CNE-2 cells were lysed in cell lysis buffer containing PMSF for 30 min at 4 °C. Lysates were collected by centrifugation at 13000 rpm for 20 min at 4 °C. Proteins from cell lysates were separated on the SDS-PAGE and transferred onto PVDF membrane (Immobion-P Transfer Membrane, Millipore Corp., Billerica, MA, USA). The membrane was blocked with PBST containing 5% non-fat dry milk for 1 h and further incubated with monoclonal anti-human HIF-1α or P-gp antibody (Santa Cruz Biotechnology, USA) overnight at 4 °C under gentle shaking. After that, the membrane was incubated with the secondary antibody (1:2000) for 2 h at room temperature. All protein bands were visualized by chemiluminescence using an ECL detection kit. Anti-β-actin antibody was used as loading control.
2.9 Cell viability study
Cytotoxicities of siRNA loaded CNPs, cisplatin and the combined use of them were determined by MTT assay in CNE-2 cells. In brief, CNE-2 cells were seeded in 96-well plates (10000 cells per well) and incubated for 24 h. The cells were then treated with different formulations for 48 h. 20 μL of MTT water solution (5 mg mL−1) was added to each well and incubated for 4 h at 37 °C. After that, the medium was removed and each well was added with 100 μL of dimethyl sulfoxide (DMSO) to dissolve the MTT formazan crystals for 5 min under gentle shaking. Absorbance was recorded at 490 nm using a microplate reader and cell viability was calculated.
2.10 Annexin V-fluorescein isothiocyanate apoptosis study
CNE-2 cells were seeded in 6-well plates (3 × 105 cells per well) and incubated for 24 h. The PBS, cisplatin, blank CNPs, siRNA loaded CNPs, blank CNPs complexed with cisplatin and siRNA loaded CNPs complexed with cisplatin was added to the wells at equivalent concentration (20 μmol L−1 of cisplatin and 60 μg mL−1 of CNPs). After incubating for 24 h, the cells were stained with annexin V-fluorescein isothiocyanate and propidium iodine staining assay kit (Invitrogen) according to the manufacturer's protocol and measured by flow cytometry (BD Accuri C6, USA).2.11 Statistical analyses
Data were provided as the mean ± standard deviation (SD) of three independent experiments. The results were analyzed by student's t-test, and one-way analysis of variance (ANOVA). P < 0.05 was considered to be statistical significant. Statistical analyses were carried out using SPSS 16.0 software (Chicago, IL, USA).3. Results
3.1 Characterization of CNPs
The effect of chitosan modification on particle size and PDI was detected by DLS. With the increasing concentration of chitosan, chitosan modification increased particle size from 246.4 ± 12.4 nm to 316.7 ± 20.5 nm and the mean PDI were less than 0.2 for all prepared formulations. Surface modification of the TPGS-b-(PCL-ran-PGA) NPs significantly changed the zeta potential from negative (non-modification) to positive and improved the siRNA EE and LE (Table 1). As Fig. 1 shows, the CNPs were spheres and uniform as revealed by the TEM micrographs. Based on the particle size, siRNA EE and LE, we used the optimal chitosan concentration of 0.8 mg mL−1 for the following experiment.| Chitosan concentration (mg mL−1) | Mean particle size (nm) | PDI | Zeta potential (mV) | siRNA EE (%) | siRNA LE (μg mg−1) |
|---|---|---|---|---|---|
| a PDI = poly dispersive index, EE = encapsulation efficiency, LE = loading efficiency, the nanoparticles were suspended in DEPC-treated PBS (pH = 7.4), error bars represent standard deviation (SD) for n = 3. | |||||
| 0 | 246.4 ± 12.14 | 0.146 ± 0.02 | −24.5 ± 0.51 | 71.1 ± 2.12 | 0.94 ± 0.03 |
| 0.2 | 276.3 ± 15.11 | 0.171 ± 0.01 | +1.84 ± 0.35 | 75.8 ± 2.56 | 1.01 ± 0.03 |
| 0.4 | 279.7 ± 14.32 | 0.188 ± 0.05 | +18.0 ± 1.41 | 77.9 ± 1.69 | 1.03 ± 0.02 |
| 0.8 | 287.3 ± 13.11 | 0.157 ± 0.06 | +30.7 ± 3.80 | 87.6 ± 0.78 | 1.16 ± 0.01 |
| 1.6 | 316.7 ± 20.52 | 0.177 ± 0.03 | +35.4 ± 0.64 | 81.1 ± 1.21 | 1.07 ± 0.02 |
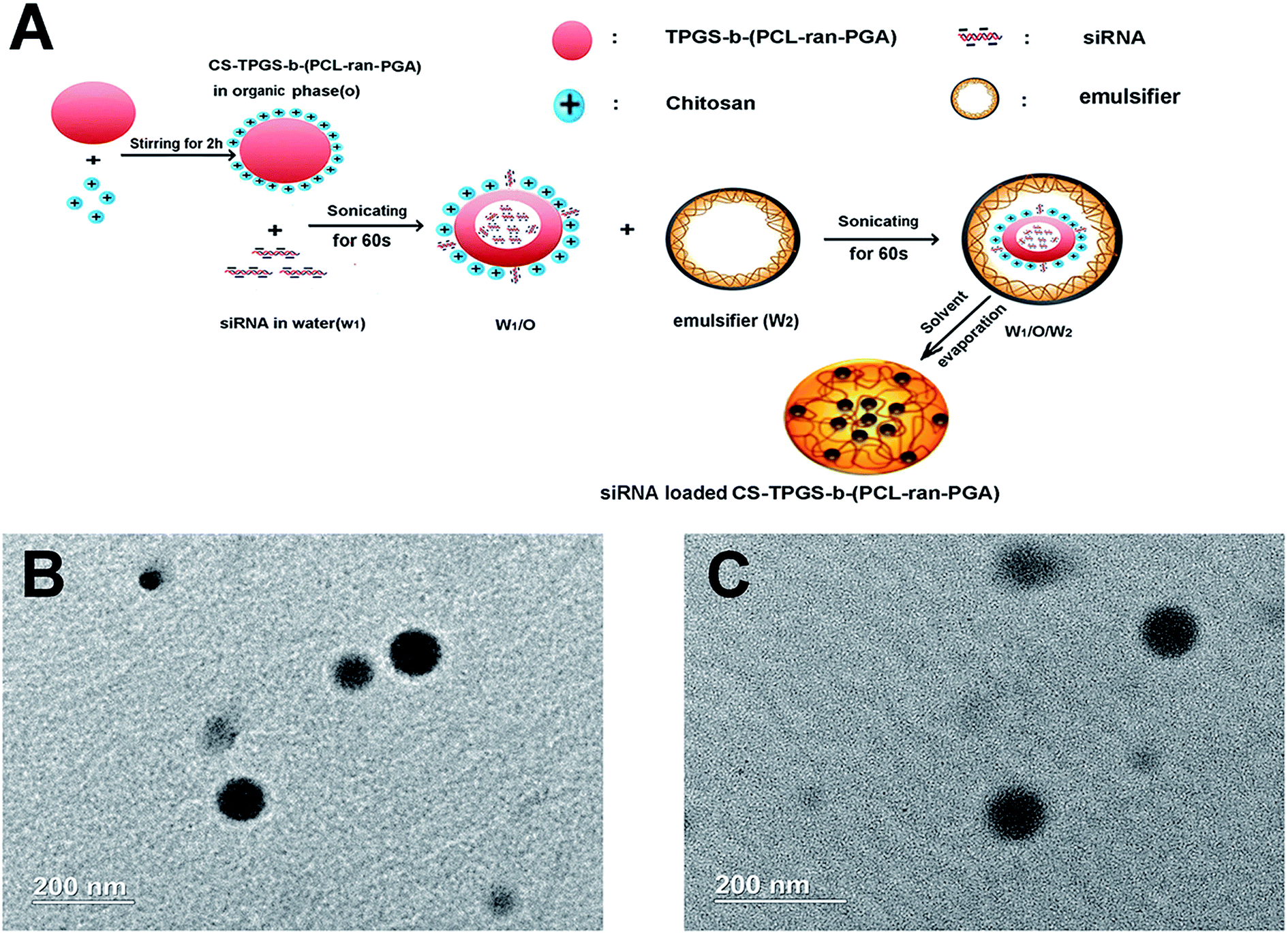 | ||
| Fig. 1 (A) Schematic representation of technique for preparation of the siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs; (B) TEM image of blank chitosan modified TPGS-b-(PCL-ran-PGA) NPs and (C) siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs. | ||
3.2 In vitro release of siRNA from CNPs
The in vitro release profile of siRNA was determined by measuring UV absorption at 260 nm at specific time points. As shown in Fig. 2, the cumulative release rates of siRNA in TE buffer over the period of 24 hours at pH 7.4 and pH 4.0 were 35.85 ± 2.53% and 44.32 ± 3.15%, respectively. The siRNAs were gradually released over the next 72 hours. The data showed that at 96 h post incubation, 62.61 ± 2.95% of siRNA released from CNPs at pH 7.4 and 81.32 ± 1.86% at pH 4.0. The release rate of siRNA from CNPs at pH 4.0 was more rapid than that at pH 7.4 (P < 0.05).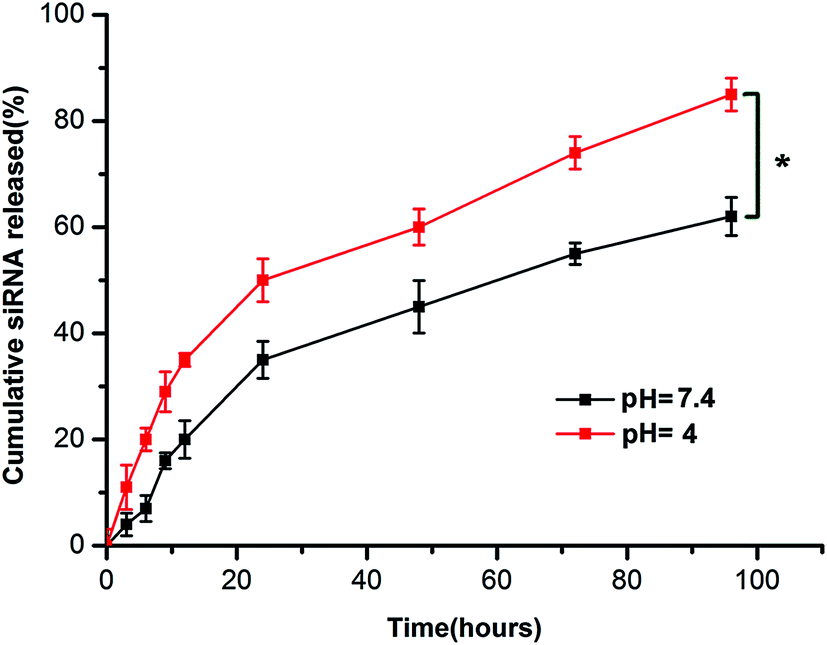 | ||
| Fig. 2 In vitro release profile of siRNA from chitosan modified TPGS-b-(PCL-ran-PGA) NPs at pH = 7.4 and pH = 4.0. Data are given as the mean ± SD (n = 3). *P < 0.05. | ||
3.3 CLSM observation of the CNPs
There was no obvious fluorescence intensity of FAM-siRNA in those cells treated with naked FAM-siRNA. A stronger fluorescent signal was observed in those cells incubated with the FAM-siRNA loaded CNPs in comparison with the cells incubated with the FAM-siRNA loaded NPs (Fig. 3). As depicted in Fig. 3I, the fluorescence of the FAM-siRNA (green) were distributed throughout the cytoplasm and closely located around the nuclei (blue, stained by DAPI) in those cells with FAM-siRNA loaded CNPs transfection.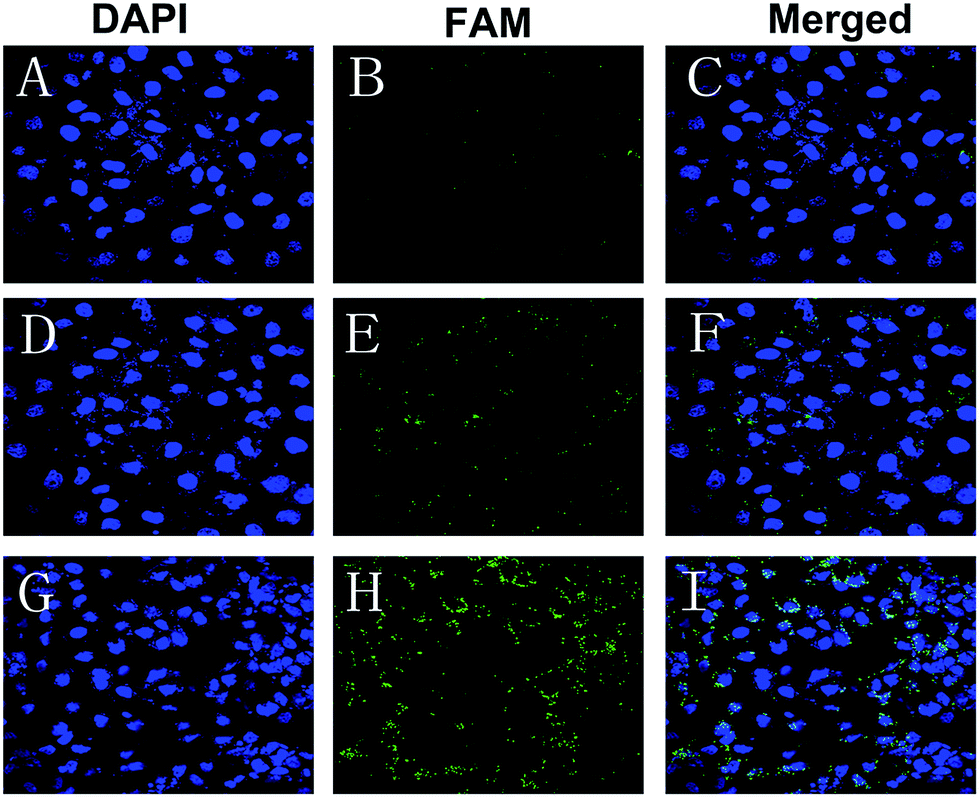 | ||
| Fig. 3 Confocal laser scanning microscopy images of CNE-2 cells transfected with the (A–C) naked FAM-siRNA, (D–F) FAM-siRNA loaded TPGS-b-(PCL-ran-PGA) NPs, and (G–I) FAM-siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs. Green color: FAM-labeled siRNAs. Blue color: cell nuclei stained by DAPI. | ||
3.4 Effect of siRNA loaded CNPs on CNE-2 cells
The RNAi effect on the expression levels of HIF-1α mRNA and protein was determined by real time RT-PCR and western blot, respectively. CNE-2 cells were transfected with naked siRNA targeting HIF-1α, siRNA targeting HIF-1α loaded NPs, or siRNA targeting HIF-1α loaded CNPs and those without any treatment were served as the control. As shown in Fig. 4, the data revealed that down-regulation efficiency of siRNA targeting HIF-1α loaded CNPs was higher than siRNA targeting HIF-1α loaded NPs transfection and for the former, the expression level of HIF-1α mRNA decreased approximately 73.43% compared with the control group. The result obtained by the western bolt assay was consistent with the real time RT-PCR result. In comparison of the control, the expression levels of HIF-1α proteins were 44.41% and 29.42% in the cells treated with siRNA targeting HIF-1α loaded NPs and siRNA targeting HIF-1α loaded CNPs, respectively.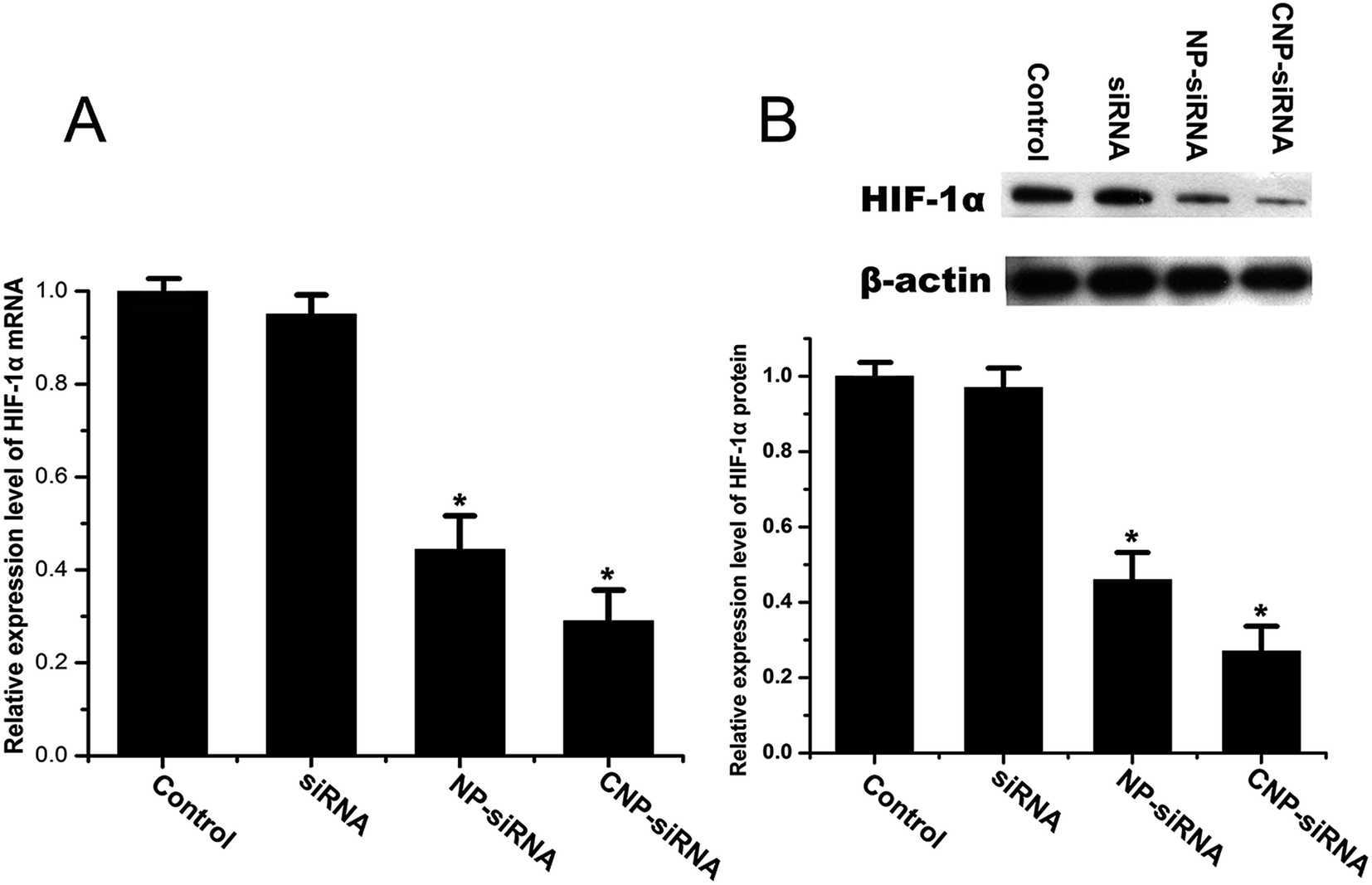 | ||
| Fig. 4 Expression level of HIF-1α determined by (A) real time RT-PCR and (B) western blot in CNE-2 cells transfected with the naked siRNA targeting HIF-1α, siRNA targeting HIF-1α loaded TPGS-b-(PCL-ran-PGA) NPs (NP-siRNA), and siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs (CNP-siRNA) under hypoxia. The HIF-1α mRNA and protein levels were normalized using β-actin mRNA and protein levels, respectively. Data are given as the mean ± SD (n = 3). *P < 0.05 as compared with the control group. | ||
Fig. 5 illustrates the cytotoxicities of different formulations in various concentrations evaluated by the MTT assay. Cells treated with the concentration of 0 μg mL−1 were considered as control. For the naked siRNA transfection, there was no significant cytotoxicity for CNE-2 cells. Scrambled siRNA loaded CNPs and blank CNPs both had a slight cytotoxic effect on the CNE-2 cells with the increasing concentrations of NPs. In comparison of the control, the cell viability was significantly decreased when transfected with siRNA targeting HIF-1α loaded CNPs with the concentration greater than 50 μg mL−1 (P < 0.05).
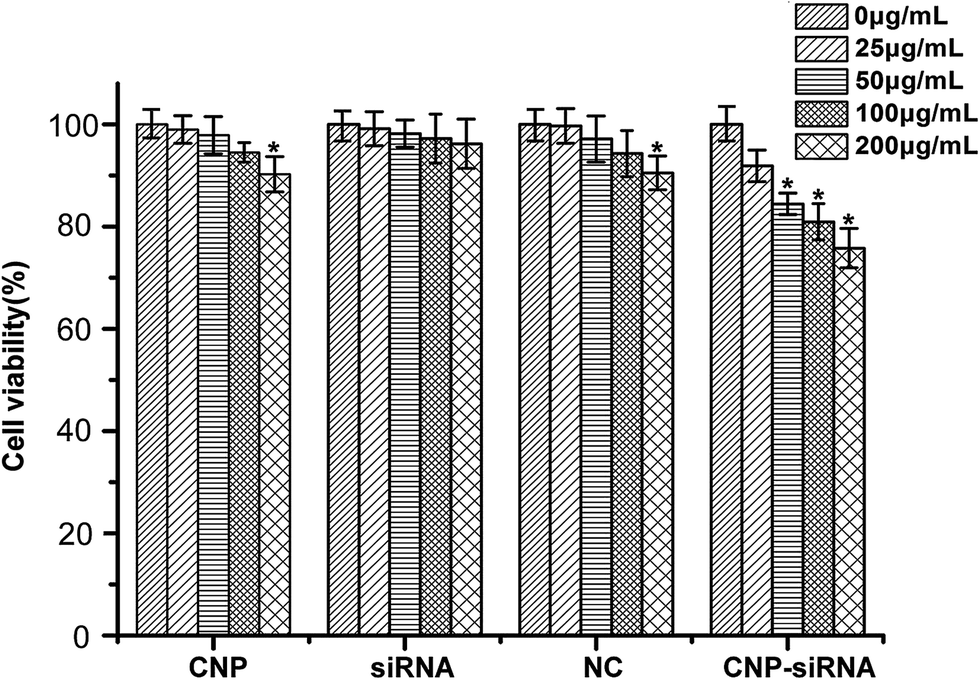 | ||
| Fig. 5 In vitro cytotoxicity of different concentrations of blank chitosan modified TPGS-b-(PCL-ran-PGA) NPs (CNP), siRNA targeting HIF-1α, scrambled siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs (NC) and siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs (CNP-siRNA) after incubation 48 h in CNE-2 cells under hypoxia. Data are given as the mean ± SD (n = 3). *P < 0.05 as compared with 0 μg mL−1 group. | ||
3.5 Chemotherapy sensitivity of cisplatin for CNE-2 cells under hypoxia
The chemotherapy sensitivity of cisplatin for CNE-2 cells were evaluated by IC50 values (concentration required to inhibit 50% of cell growth). CNE-2 cells were seeded in 96-well plates and treated with cisplatin alone under normoxic or hypoxic condition, cisplatin in combination with scrambled siRNA loaded CNPs or siRNA targeting HIF-1α loaded CNPs under hypoxic condition where the cisplatin concentration ranged from 0 to 36 μmol L−1 and the siRNA dose was 50 nM.As Fig. 6 shows, the IC50 value of cisplatin was found to be 20.18 μmol L−1 in CNE-2 cells with cisplatin treatment alone under normoxic condition but increased to be 33.97 μmol L−1 when exposed to hypoxia condition. With respect to the combination treatment of scrambled siRNA loaded CNPs and cisplatin in hypoxic circumstance, the IC50 value of cisplatin was 27.28 μmol L−1. Whereas the IC50 value was decreased to 11.55 μmol L−1 when the cells were treated with siRNA targeting HIF-1α loaded CNPs and cisplatin in combination treatment under hypoxic condition.
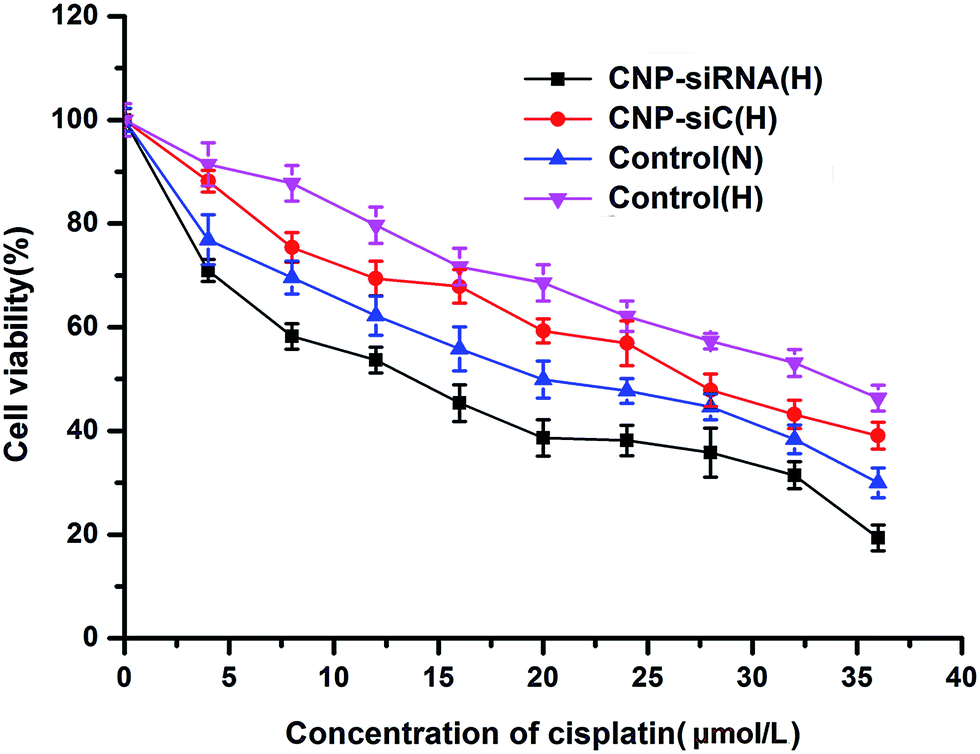 | ||
| Fig. 6 In vitro cytotoxicity of different concentrations of cisplatin after incubation 48 h in CNE-2 cells under normoxic and hypoxic conditions. CNP-siRNA(H): cells treated with siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs complexed with cisplatin under hypoxic condition; NC(H): cells treated with scrambled siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs complexed with cisplatin under hypoxic condition; control(N): cells treated with cisplatin under normoxic condition; control(H): cells treated with cisplatin under hypoxic condition. | ||
To explore the potential molecular mechanisms of chemoresistance of cisplatin for CNE-2 cells under hypoxia, the expression levels of HIF-1α and its target gene MDR1 as well as MDR1 encoded P-gp were detected. The expression levels of HIF-1α and MDR1/P-gp in CNE-2 cells were significantly increased after incubation for 24 h in hypoxia compared with those in normoxic circumstance (Fig. 7). By treatment with siRNA targeting HIF-1α loaded CNPs, HIF-1α and MDR1/P-gp expression levels were reduced significantly in comparison with the scrambled siRNA loaded CNPs treatment.
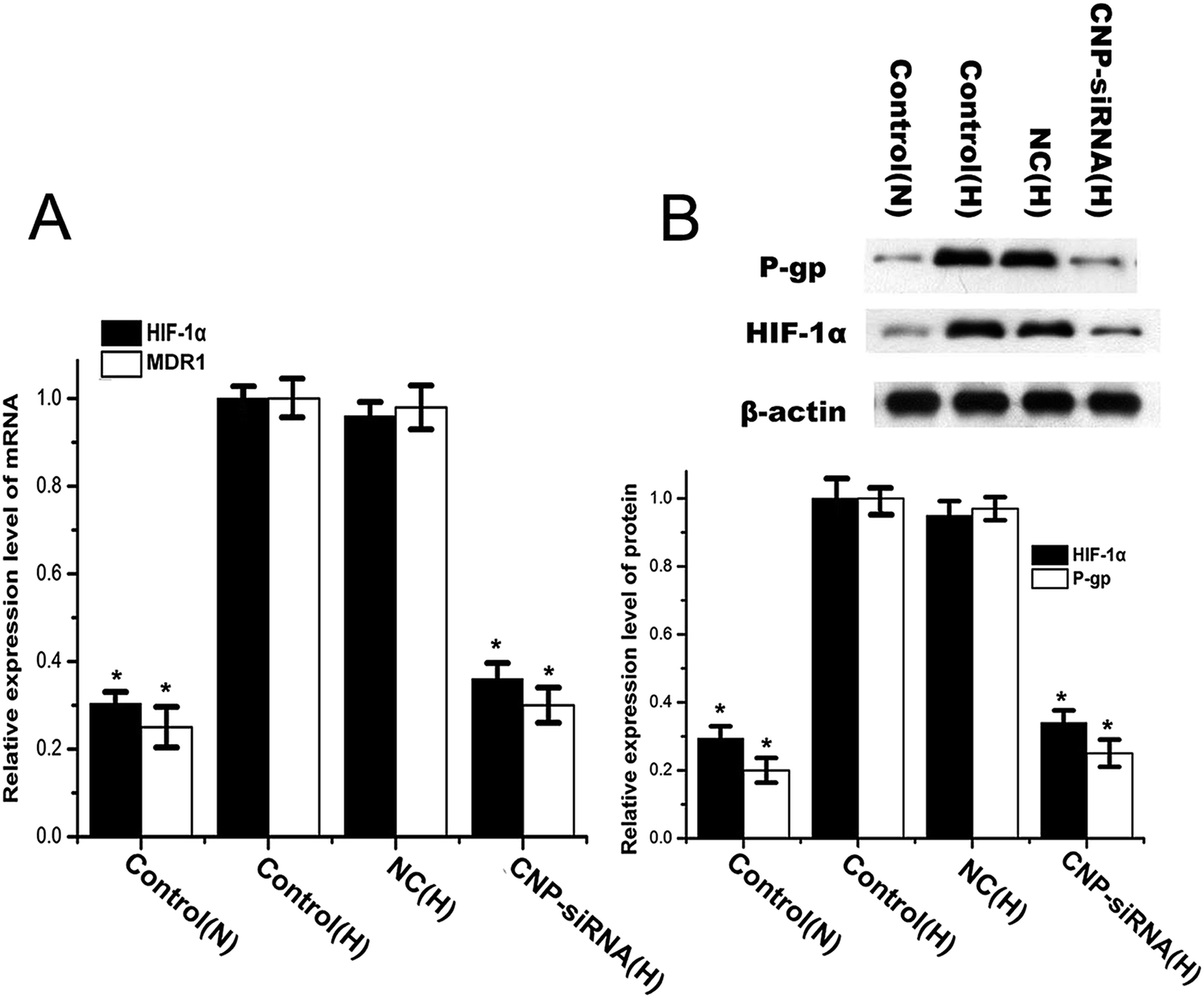 | ||
| Fig. 7 Expression levels of HIF-1α and MDR1/P-gp determined by (A) real time RT-PCR and (B) western blot in CNE-2 cells under normoxic and hypoxic conditions. Control(N): cells treated with cisplatin under normoxic condition; control(H): cells treated with cisplatin under hypoxic condition; NC(H): cells treated with scrambled siRNA loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs complexed with cisplatin under hypoxic condition; CNP-siRNA(H): cells treated with siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs complexed with cisplatin under hypoxic condition. The mRNA and protein levels of HIF-1α and MDR1/P-gp were normalized using β-actin mRNA and protein levels, respectively. Data are given as the mean ± SD (n = 3). *P < 0.05 as compared with the control(H) group. | ||
3.6 Synergistic anticancer effect of siRNA loaded CNPs and cisplatin
To evaluate the synergistic anticancer effect of siRNA loaded CNPs and cisplatin, CNE-2 cells were treated with PBS, cisplatin alone, blank CNPs, siRNA nanoformulation, blank CNPs complexed with cisplatin and siRNA nanoformulation complexed with cisplatin containing equivalent drug concentration (20 μmol L−1 of cisplatin and 60 μg mL−1 of CNPs) for 48 h. MTT assay revealed the corresponding cell viability was 98.3 ± 0.032%, 97.12 ± 1.11%, 68.12 ± 4.23%, 55.54 ± 3.63%, 84.31 ± 3.11%, and 38.23 ± 3.43%, respectively (Fig. 8A). Annexin V-fluorescein isothiocyanate apoptosis study also showed that siRNA nanoformulation complexed with cisplatin induced more cellular apoptosis in CNE-2 cells compared to the other treatments (Fig. 8B). These results indicated that synergistic anticancer activities could be obtained by siRNA loaded CNPs and cisplatin in combination.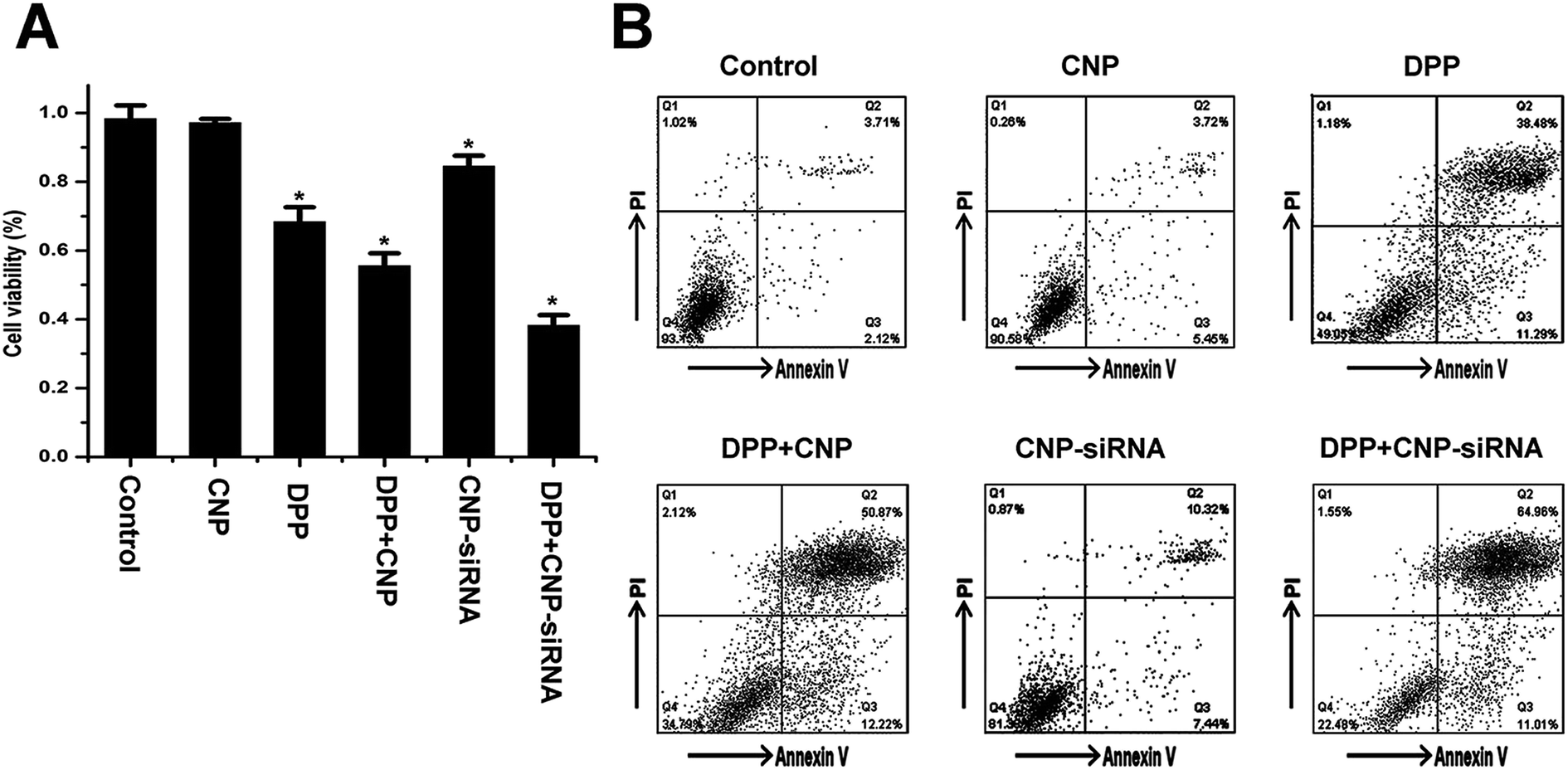 | ||
| Fig. 8 In vitro cytotoxicity of different formulations after incubation 48 h in CNE-2 cells under hypoxia determined by (A) MTT assay and (B) flow cytometry. CNP: blank chitosan modified TPGS-b-(PCL-ran-PGA) NPs; CNP-siRNA: siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs; DDP, cisplatin. Data are given as the mean ± SD (n = 3). *P < 0.05 as compared with the PBS control group. | ||
As shown in Fig. 9, HIF-1α mRNA and protein levels in cells treated with the siRNA loaded CNPs were significantly decreased compared with the PBS control group. HIF-1α mRNA and protein levels were found to be further reduced after treatment with the combination of siRNA loaded CNPs and cisplatin. Similar results were observed in the expression of MDR1 and P-gp. No significant differences in HIF-1α and MDR1/P-gp expression levels were noted between the PBS, cisplatin, blank CNPs, blank CNPs complexed with cisplatin treatment groups.
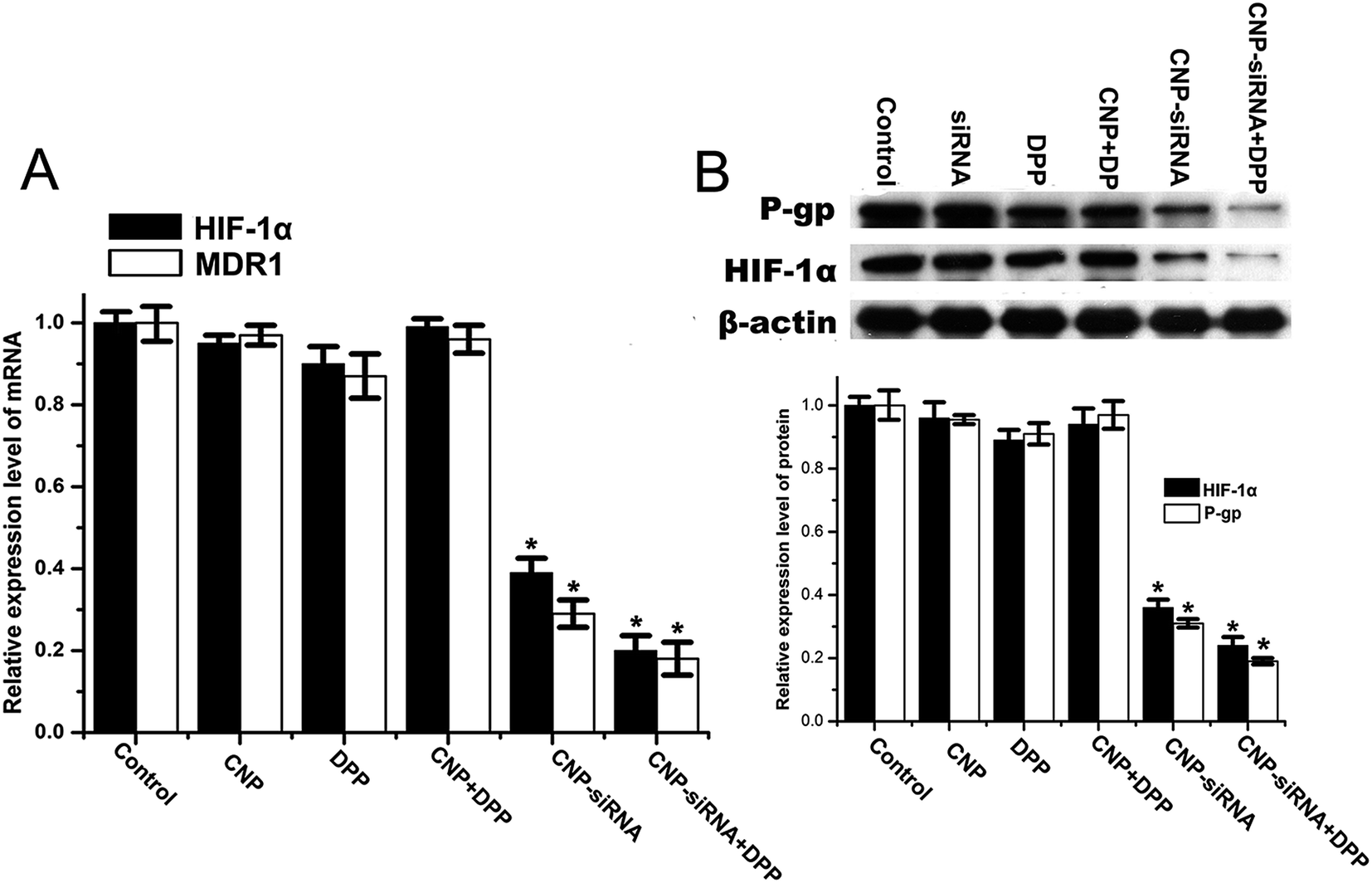 | ||
| Fig. 9 Expression levels of HIF-1α and MDR1/P-gp determined by (A) real time RT-PCR and (B) western blot in CNE-2 cells with different formulations treatment under hypoxia. CNP: blank chitosan modified TPGS-b-(PCL-ran-PGA) NPs; CNP-siRNA: siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs; DDP, cisplatin. The mRNA and protein levels of HIF-1α and MDR1/P-gp were normalized using β-actin mRNA and protein levels, respectively. *P < 0.05 as compared with the PBS control group. | ||
4. Discussion
Lots of evidences have demonstrated that hypoxic microenvironment contributes to the carcinogenesis and development of cancer cells.25 Moreover, hypoxia condition induces tumor cells with series of genetic and metabolic changes to maintain resistance to anticancer drugs.26 Since HIF-1α is an important factor in the adaptation of cells to hypoxic environment, many studies pay much attention to investigate the HIF-1α related signaling pathways mediating chemotherapy resistance and put much efforts to develop different approaches to attenuate the biological functions of HIF-1α and its downstream genes in cancer cells. Thus, in this context, we prepared a chitosan modified TPGS-b-(PCL-ran-PGA) NPs to deliver siRNA targeting HIF-1α into CNE-2 cells to inhibit the expression of HIF-1α and enhance the treatment efficacy of cisplatin for NPC.In recent years, many biodegradable and biocompatible NPs are applied as the delivery systems for anticancer agents.27 While the hindrance for most NPs is the negative surface charge, which may limit their cellular uptake due to negative surface charge of the cell membrane. Currently the chitosan modification is widely adopted to neutralize the negative surface charge of NPs. Hence we used cationic chitosan for coating the outer surface of TPGS-b-(PCL-ran-PGA) NPs, resulting in the zeta potential shifted from negative (non-modification) to positive. In accordance with previous report, the diameter of CNPs detected by DLS experiment was relatively larger than that by TEM study, which might be ascribed to that NPs in dry circumstance are inclined to shrink and collapse.28 In this experiment, we found that when NPs were modified with a chitosan concentration of 0.8 mg mL−1, the diameter of NPs was less than 300 nm without aggregation and the siRNA EE was highest. It has been reported that the size of biodegradable NP for gene delivery to be less than 300 nm would be conducive to increasing the encapsulation efficiency of gene and promoting the cellular uptake of gene loaded NPs.29 Thus, the chitosan concentration of 0.8 mg mL−1 was used for the further study. The double emulsion solvent evaporation method was used for the encapsulation of siRNA into the inner core of NPs in our previous study and achieved a siRNA EE around 70%.23 However, those unencapsulated siRNAs were lost during the NP prepare procedure. By using the chitosan modified onto the surface of NPs, with the same fabrication method, some unencapsulated siRNAs could be absorbed onto the cationic surface of chitosan layer and the siRNA EE was significantly improved. The siRNA EE of NPs affects the silencing effectiveness of the target gene and its consequent biological effects on the tumor development.
It is essential for a delivery carrier to facilitate drug release from formulation with a steady and constant rate over a considerable period. The controlled release property of the present CNPs was observed over a period of 96 h and there existed a reduction of the burst release in a short period of time in the chitosan modified formulation compared with the unmodified one at pH 7.4 (data not shown). As shown in Fig. 2, the siRNA release ratio at pH 4.0 was higher than that at pH 7.4, which might attributed to the acidification of the microenvironment induced deformation of the core–shell structure of the NPs. The in vitro siRNA release results indicated that the CNPs could approach accelerated release of entrapped siRNA for gene therapy in the acidic microenvironment of tumor cells.
It was reported that NPs reach to the cytosol through escaping the endosomal degradation pathway after entering the cells.30 The current in vitro cellular uptake experiment showed that the fluorescent of FAM-siRNA could be internalized into the cells and closely located around the nuclei, indicating that the CNPs could successfully transfect siRNA into cytoplasm following endosomal escape. More FAM-siRNA loaded CNPs were taken up by CNE-2 cells than FAM-siRNA loaded NPs in the present study, suggesting that CNPs could serve as an appropriate delivery carrier for promoting siRNA absorption. Thus, it is reasonable to assume that siRNA released from the present CNPs was able to conduct gene inhibition or knockdown activity. The real time RT PCR and western blot results confirmed this assumption. The expression levels of HIF-1α mRNA and protein in cells treated with siRNA targeting HIF-1α loaded CNPs were significantly decreased compared with control cells. HIF-1α regulates several downstream genes expression, such as epidermal growth factor receptor, BH3 interacting domain death agonist, glucose transporter-1, and DNA double-strand break repair enzymes, to participate in the process of cancer progression and apoptosis inhibition.31–34 Decrease in HIF-1α expression levels may lead to increase in cell apoptosis. As the cell viability experiment showed, blank CNPs and scrambled siRNA loaded CNPs were found to have a slight cytotoxic effect on cell survival with dose increasing, which might be due to anticancer efficacy of TPGS to induce apoptosis. Anticancer growth property of TPGS has been observed in cell line and in animal models and the synergistic anticancer effects through the combinations of TPGS with other anticancer agents were well documented.35–37 Loss of viability was observed among the cells treated with siRNA targeting HIF-1α loaded CNPs, which might be attributed to synergistic anticancer capability of the TPGS and silencing effect of siRNA targeting HIF-1α as explained above.
The chemotherapy sensitivity of cisplatin for CNE-2 cells were examined in normoxia and hypoxia. IC50 value of cisplatin was significantly increased when the CNE-2 cells exposed to hypoxia condition. While the addition of siRNA targeting HIF-1α loaded CNPs restored the effect of cisplatin and IC50 value was significantly reduced under hypoxia. This finding was consistent with the previous observations that inhibiting HIF-1α can restore chemosensitivity in different tumors, such as gastric cancer, fibrosarcoma and breast carcinoma.6,38,39 The present study showed that hypoxia condition up-regulated the expression of HIF-1α and MDR1 encoded P-gp. HIF-1α inhibition by transfecting cells with siRNA targeting HIF-1α loaded CNPs significantly reduced the expression of MDR1 mRNA and P-gp. HIF-1α mediated MDR1/P-gp expression contributes to hypoxia-induced drug resistance because P-gp transporter of tumor cells may actively pump the cytotoxic drugs out to reduce their intracellular accumulation and thereby alleviate their antitumor effects.40,41 With respect to the therapeutic effect of different formulation under hypoxia, the CNE-2 cells were treated with free cisplatin, blank CNPs, siRNA nanoformulation, blank CNPs complexed with cisplatin and siRNA nanoformulation complexed with cisplatin. As Fig. 8 shows, the free cisplatin, siRNA nanoformulation and blank CNPs complexed with cisplatin demonstrated reducing effects on cell viability. Interestingly, the blank CNPs could enhance the sensitivity of free cisplatin for CNE-2 cells, indicating some other molecular mechanisms were underlying since the HIF-1α and MDR1/P-gp expression levels had no significant differences compared with the control. This might be ascribed to the capability of TPGS to improve drug permeability. TPGS has been found to induce ATPase activity inhibition to reduce P-gp mediated efflux and thus increase the absorption of drug to reverse multidrug resistance of tumor cells.42,43 Significant increase of cytotoxicity on CNE-2 cells was observed when treated with siRNA nanoformulation complexed with cisplatin, which suggested that siRNA loaded CNPs in combination with cisplatin can exert the synergistic anticancer effects. Additionally, with the combination therapy, cisplatin can achieve obvious cytotoxicity in a lower concentration as the IC50 value revealed in the present in vitro study. Further studies will focus on the therapeutic efficacy and side effects of cisplatin in combination with siRNA nanoformulation in vivo.
5. Conclusion
In this in vitro study, chitosan was adopted for cationic surface modification of TPGS-b-(PCL-ran-PGA) NPs. The results revealed that chitosan-modified TPGS-b-(PCL-ran-PGA) NPs could efficiently deliver siRNA into CNE-2 cells to down-regulate the expression of HIF-1α and decrease the cell viability. Using siRNA loaded chitosan-modified TPGS-b-(PCL-ran-PGA) NPs to reduce HIF-1α mediated P-gp expression significantly enhanced the cisplatin sensitivity of CNE-2 cells under hypoxia. Synergistic antitumor activities could be obtained by the use of combinations of siRNA targeting HIF-1α loaded chitosan modified TPGS-b-(PCL-ran-PGA) NPs and cisplatin. Chitosan-modified TPGS-b-(PCL-ran-PGA) NPs can serve as an optimizing carrier for siRNA delivery and provide potential to increase the therapeutic efficacy of chemotherapeutic drugs.Conflict of interest
The authors report no conflicts of interest in this work.Acknowledgements
This work was supported by Special Fund for the development of strategic emerging industries of Shenzhen, China (No. JCYJ20120830162655505).References
- Y. Wang, W. Ding, C. Chen, Z. Niu, M. Pan and H. Zhang, J. Cancer Res. Ther., 2015, 11(2), C191–C195 Search PubMed.
- X. Ou, X. Zhou, Q. Shi, X. Xing, Y. Yang, T. Xu, C. Shen, X. Wang, X. He, L. Kong, H. Ying and C. Hu, OncoTargets Ther., 2015, 6, 38381–38397 Search PubMed.
- R. C. Ji, Cancer Lett., 2014, 346, 6–16 CrossRef CAS PubMed.
- Y. Chen, X. Li, S. Wu, G. Xu, Y. Zhou, L. Gong, Z. Li and D. Yang, Med. Oncol., 2014, 31, 304 CrossRef PubMed.
- E. B. Rankin and A. J. Giaccia, Cell Death Differ., 2008, 15, 678–685 CrossRef CAS PubMed.
- L. Liu, X. Ning, L. Sun, H. Zhang, Y. Shi, C. Guo, S. Han, J. Liu, S. Sun, Z. Han, K. Wu and D. Fan, Cancer Sci., 2008, 99, 121–128 CAS.
- D. W. Li, P. Dong, F. Wang, X. W. Chen, C. Z. Xu and L. Zhou, Asian Pac. J. Cancer Prev., 2013, 14, 4853–4858 CrossRef PubMed.
- J. Adamski, A. Price, C. Dive and G. Makin, PLoS One, 2013, 8, e65304 CAS.
- X. Song, X. Liu, W. Chi, Y. Liu, L. Wei, X. Wang and J. Yu, Cancer Chemother. Pharmacol., 2006, 58, 776–784 CrossRef CAS PubMed.
- X. P. Sun, X. Dong, L. Lin, X. Jiang, Z. Wei, B. Zhai, B. Sun, Q. Zhang, X. Wang, H. Jiang, G. W. Krissansen, H. Qiao and X. Sun, FEBS J., 2014, 281, 115–128 CrossRef CAS PubMed.
- S. Cipro, J. Hrebackova, J. Hrabeta, J. Poljakova and T. Eckschlager, Oncol. Rep., 2012, 27, 1219–1226 CAS.
- Y. Shan, X. Li, B. You, S. Shi, Q. Zhang and Y. You, Oncol. Rep., 2015, 34, 1943–1952 Search PubMed.
- T. Tokatlian and T. Segura, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 305–315 CrossRef CAS PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–U1140 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- T. Wang, D. Zhu, G. Liu, W. Tao, W. Cao, L. Zhang, L. Wang, H. Chen, L. Mei, L. Huang and X. Zeng, RSC Adv., 2015, 5, 50617–50627 RSC.
- E. Bilensoy, Expert Opin. Drug Delivery, 2010, 7, 795–809 CrossRef CAS PubMed.
- M. L. Tan, P. F. Choong and C. R. Dass, J. Pharm. Pharmacol., 2009, 61, 3–12 CrossRef CAS PubMed.
- L. Chronopoulou, M. Massimi, M. F. Giardi, C. Cametti, L. C. Devirgiliis, M. Dentini and C. Palocci, Colloids Surf., B, 2013, 103, 310–317 CrossRef CAS PubMed.
- K. H. Min, K. Park, Y. S. Kim, S. M. Bae, S. Lee, H. G. Jo, R. W. Park, I. S. Kim, S. Y. Jeong, K. Kim and I. C. Kwon, J. Controlled Release, 2008, 127, 208–218 CrossRef CAS PubMed.
- E. N. Koukaras, S. A. Papadimitriou, D. N. Bikiaris and G. E. Froudakis, RSC Adv., 2014, 4, 12653 RSC.
- H. V. Jagani, V. R. Josyula, V. R. Palanimuthu, R. C. Hariharapura and S. S. Gang, Eur. J. Pharm. Sci., 2013, 48, 611–618 CrossRef CAS PubMed.
- Y. Chen, G. Xu, Y. Zheng, M. Yan, Z. Li, Y. Zhou, L. Mei and X. Li, Int. J. Nanomed., 2015, 10, 1375–1386 CrossRef CAS PubMed.
- D. K. Jensen, L. B. Jensen, S. Koocheki, L. Bengtson, D. Cun, H. M. Nielsen and C. Foged, J. Controlled Release, 2012, 157, 141–148 CrossRef CAS PubMed.
- C. M. Tang and J. Yu, J. Gastroenterol. Hepatol., 2013, 28, 401–405 CrossRef CAS PubMed.
- H. Doktorova, J. Hrabeta, M. A. Khalil and T. Eckschlager, Biomed. Pap., 2015, 159, 166–177 Search PubMed.
- A. P. Subramanian, S. K. Jaganathan and E. Supriyanto, RSC Adv., 2015, 5, 72638–72652 RSC.
- X. Zeng, W. Tao, L. Mei, L. Huang, C. Tan and S. S. Feng, Biomaterials, 2013, 34, 6058–6067 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoter, H. Epstein-Barash, L. G. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–U102 CrossRef CAS PubMed.
- N. Akakura, M. Kobayashi, I. Horiuchi, A. Suzuki, J. X. Wang, J. Chen, H. Niizeki, K. Kawamura, M. Hosokawa and M. Asaka, Cancer Res., 2001, 61, 6548–6554 CAS.
- A. Unruh, A. Ressel, H. G. Mohamed, R. S. Johnson, R. Nadrowitz, E. Richter, D. M. Katschinski and R. H. Wenger, Oncogene, 2003, 22, 3213–3220 CrossRef CAS PubMed.
- J. T. Erler, C. J. Cawthorne, K. J. Williams, M. Koritzinsky, B. G. Wouters, C. Wilson, C. Miller, C. Demonacos, I. J. Stratford and C. Dive, Mol. Cell. Biol., 2004, 24, 2875–2889 CrossRef CAS PubMed.
- H. P. Gerber, A. McMurtrey, J. Kowalski, M. H. Yan, B. A. Keyt, V. Dixit and N. Ferrara, J. Biol. Chem., 1998, 273, 30336–30343 CrossRef CAS PubMed.
- H. J. Youk, E. Lee, M. K. Choi, Y. J. Lee, J. H. Chung, S. H. Kim, C. H. Lee and S. J. Lim, J. Controlled Release, 2005, 107, 43–52 CrossRef CAS PubMed.
- J. Neuzil, M. Tomasetti, Y. Zhao, L.-F. Dong, M. Birringer, X.-F. Wang, P. Low, K. Wu, B. A. Salvatore and S. J. Ralph, Mol. Pharmacol., 2007, 71, 1185–1199 CrossRef CAS PubMed.
- C. Constantinou, A. Papas and A. I. Constantinou, Int. J. Cancer, 2008, 123, 739–752 CrossRef CAS PubMed.
- J. Hao, X. Song, B. Song, Y. Liu, L. Wei, X. Wang and J. Yu, Cancer Gene Ther., 2008, 15, 449–455 CrossRef CAS PubMed.
- X. L. Dong, P. F. Xu, C. Miao, Z. Y. Fu, Q. P. Li, P. Y. Tang and T. Wang, Biomed. Pharmacother., 2012, 66, 70–75 CrossRef CAS PubMed.
- K. M. Comerford, T. J. Wallace, J. Karhausen, N. A. Louis, M. C. Montalto and S. P. Colgan, Cancer Res., 2002, 62, 3387–3394 CAS.
- A. K. Tiwari, K. Sodani, C. L. Dai, C. R. Ashby Jr and Z. S. Chen, Curr. Pharm. Biotechnol., 2011, 12, 570–594 CAS.
- E.-M. Collnot, C. Baldes, M. F. Wempe, R. Kappl, J. Huettermann, J. A. Hyatt, K. J. Edgar, U. F. Schaefer and C.-M. Lehr, Mol. Pharm., 2007, 4, 465–474 CrossRef CAS PubMed.
- Z. Zhang, L. Mei and S.-S. Feng, Nanomedicine, 2012, 7, 1645–1647 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |