
Systems metabolic engineering of Escherichia coli to enhance the production of flavonoid glucuronides†
Abstract
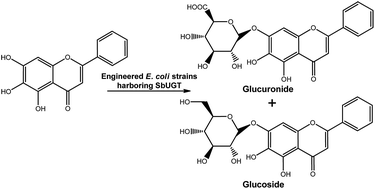
Large-scale application of whole-cell glycosylation is still hampered by inefficient UDP-sugar formation. Using a module-based approach, an engineered Escherichia coli strain was constructed to enhance the production of flavonoid glucuronides. The engineered metabolic pathway was partitioned into two modules: an endogenous upstream biosynthetic pathway to produce the sugar donor UDP-glucuronic acid (UDPGA) and a heterologous downstream UDP-dependent glycosyltransferase (UGT) to catalyse the glucuronidation of flavonoids. First, two UGTs (SbUGT-W76 and SbUGT-W112) were isolated from Scutellaria baicalensis Georgi. Enzymatic assays showed that the recombinant SbUGT exhibited regiospecificity towards the C7-OH position of flavonoid substrates, and conflicting results on the specificity of sugar donor and acceptor compared with that of reported SbUBGAT were obtained. Then, the native upstream module of the UDPGA synthetic pathway was strengthened to increase the endogenous level of UDPGA. Using these strategies, up to 797 mg L−1 baicalein-7-O-glucuronide was biosynthesized. Furthermore, systemic engineering of upstream and downstream genes in the glucuronide biosynthetic pathway was conducted for characterization and the capability to biotransform baicalein. The results showed that UDP-glucose 6-dehydrogenase (Ugd) catalysed the rate-determining step for glucuronide production. SbUGT displayed the same catalytic properties both in vivo and in vitro.
 Please wait while we load your content...
Please wait while we load your content...

