
Atom-economical synthesis of 3,3,3-trifluoropropanal dialkyl acetals through Pd/C catalyzed acetalization of 3,3,3-trifluoropropene†
Abstract
A facile and efficient procedure for one-step synthesis of 3,3,3-trifluoropropanal dialkyl acetals from readily available 3,3,3-trifluoropropene (TFP) has been developed. The catalyst can be recycled for 4 times without obvious deactivation. This process provides a novel and atom-economical synthetic strategy for the preparation of functional CF3-containing compounds.
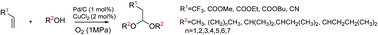
 Please wait while we load your content...
Please wait while we load your content...

