
Selective CO adsorbent CuCl/AC prepared using CuCl2 as a precursor by a facile method
Abstract
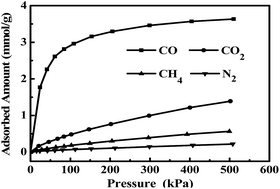
Activated carbon (AC) supported CuCl (CuCl/AC) has been prepared with CuCl2 as precursor by a monolayer dispersion method. The samples are investigated for CO adsorption and characterized by inductively coupled plasma optical emission spectrometry, X-ray diffraction, N2 adsorption/desorption and X-ray photoelectron spectroscopy. The characterization results reveal that CuCl2 supported on AC can be completely converted to CuCl after activation at 543 K in N2. The resulting adsorbent displays high CO adsorption capacity, high CO/N2, CO/CH4 and CO/CO2 adsorption selectivities and excellent reversibility, and the adsorption equilibrium isotherms of CO on the adsorbents at temperatures up to 333 K can be well fitted by both the Langmuir and Sips models.
 Please wait while we load your content...
Please wait while we load your content...

