
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Importance of the linker region in matrix metalloproteinase-1 domain interactions†
Warispreet
Singh
a,
Gregg B.
Fields
*bc,
Christo Z.
Christov
*a and
Tatyana G.
Karabencheva-Christova
*a
aDepartment of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: tatyana.karabencheva-christova@northumbria.ac.uk; christo.christov@northumbria.ac.uk
bDepartment of Chemistry & Biochemistry, Florida Atlantic University, Jupiter, FL 33458, USA. E-mail: fieldsg@fau.edu
cDepartment of Chemistry, The Scripps Research Institute/Scripps Florida, Jupiter, FL 33458, USA
First published on 24th February 2016
Abstract
Collagenolysis is catalyzed by enzymes from the matrix metalloproteinase (MMP) family, where one of the most studied is MMP-1. The X-ray crystallographic structure of MMP-1 complexed with a collagen-model triple-helical peptide (THP) provided important atomistic information, but few details on the effects of the conformational flexibility on catalysis. In addition, the role of the linker region between the catalytic (CAT) and hemopexin-like (HPX) domains was not defined. In order to reveal the dynamics and correlations of MMP-1 comprehensive atomistic molecular dynamics simulations of an MMP-1·THP complex was performed. To examine the role of the linker region for MMP-1 function simulations with linker regions from MT1-MMP/MMP-14 and MMP-13 replacing the MMP-1 linker region were performed. The MD studies were in good agreement with the experimental observation that in the MMP-1·THP X-ray crystallographic structure MMP-1 is in a “closed” conformation. MD revealed that the interactions of the THP with the both the CAT and HPX domains of MMP-1 are dynamic in nature, and the linker region of MMP-1 influences the interactions and dynamics of both the CAT and HPX domains and collagen binding to MMP-1.
Introduction
One of the primary components of the extracellular matrix (ECM) is collagen, which is also the most abundant protein in mammals.1 There are at least 29 different types of collagens that occur in vertebrates. The collagen molecule consists of three polypeptide strands (α chains) that organize themselves in a ropelike triple-helix conformation, stabilized by inter-chain hydrogen bonding. The degradation of collagen is a key process in normal development and homeostasis; however, unbalanced collagenolysis contributes to numerous pathologies such as cancer, arthritis, and cardiovascular and neurodegenerative diseases. Amongst the enzymes capable of collagen catabolism in vertebrates are matrix metalloproteinases (MMPs). MMP-1, also known as interstitial collagenase or fibroblast collagenase, is a zinc and calcium dependent endopeptidase located in the ECM in vertebrates.2–5 MMP-1 degrades interstitial (types I–III) collagen into 3/4 and 1/4 fragments.6,7 The three-dimensional X-ray crystallographic structure of catalytically inactive (E200A) MMP-1 complexed with a collagen-model triple-helical peptide (THP) has been solved by Manka et al. (Fig. 1).8 MMP-1 in this structure consists of the N-terminal catalytic (CAT) domain, the C-terminal hemopexin-like (HPX) domain, and the linker region connecting the CAT and HPX domains. The THP, with three strands designated as leading (L), middle (M), and trailing (T), is approximately 115 Å in length and makes extensive interaction with both the CAT and HPX domains.8 In concert with the X-ray crystallographic “snapshot” of MMP-1·THP, dynamic information about MMP-1 interaction with triple-helices was obtained through a series of NMR spectroscopic structures and molecular docking calculations.9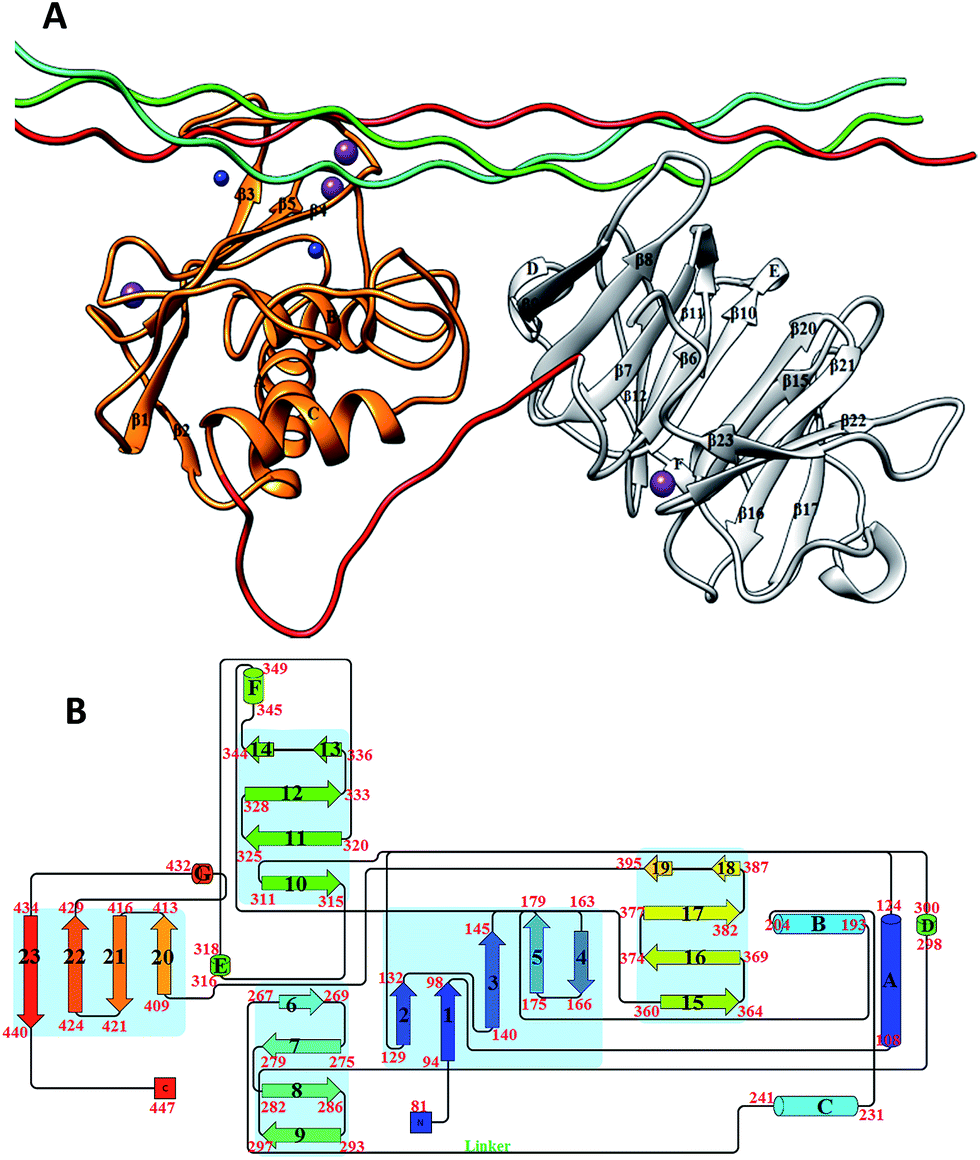 | ||
| Fig. 1 (A) The 3D structure of human MMP-1 (E200A) complexed with a THP (PDB 4AUO8) drawn using UCSF Chimera.16 MMP-1 (displayed in silhouette round ribbon) consists of the CAT domain (orange), inter-domain linker (red), and HPX domain (grey). The L, M, and T strands of the THP are shown in tube representation in cyan, green, and red, respectively. The zinc and calcium ions bound to the enzyme are shown in spherical representation in blue and purple, respectively. (B) 2D topology diagram of (E200A) MMP-1 made using Pro-origami tool using STRIDE algorithm for secondary structure prediction.17 The color gradient of blue and red are used for the N- and C-termini of the protein. The β-sheets are represented numerically and the α-helices alphabetically. | ||
Molecular dynamics (MD) simulations have been applied to study the MMP family of enzymes. Diaz and coworkers observed a conformational shift from a closed X-ray crystallographic MMP-2 structure to an elongated structure using atomistic MD simulations on a 100 ns time scale.10 MMP-2 can adopt extended conformations in solution prior to collagen hydrolysis.11 MD has provided insight into the degradation of type I collagen fibrils by MMP-8,12 but limited efforts have been made to study MMP-1. Presently, the largest collection of experimental data for MMP-catalyzed collagenolysis has been obtained using MMP-1,8,9,13–15 yet there is still much to be learned as to how conformational flexibility and dynamics influences MMP-1 structure–function relationships, and specifically to provide insight on the effects of dynamics on the interactions between the MMP-1 CAT and HPX domains and with collagen triple-helices. In addition, it is important to understand how the composition and the flexibility of the linker region influence the interactions between the MMP-1 CAT and HPX domains and with collagen. In order to provide insight into MMP-1-catalyzed collagenolysis, we herein performed a set of long-range atomistic (AT) MD simulations on an MMP-1·THP complex. Effects of the linker region on inter-domain interactions and interactions with the substrate were studied by MD simulations of in silico engineered MMP-1 with the linker of MMP-14/MT1-MMP or MMP-13.
Methods
Initial structure preparation
The coordinates of the wild type MMP-1 bound to a THP model of the type II collagen MMP cleavage site were obtained from the Protein Data Bank18 (PDB 4AUO8). SwissPDBViewer19 was used for adding missing atoms and selecting one from the alternative side chain orientations.19 The active form of MMP-1 was made by substituting the A200 with E200 using the Modeller20 program. The linker of MMP-14 and MMP-13 were modelled into the linker region of MMP-1 by using the Modeller20 program. The numbering of the THP in the MMP-1·THP complex and subsequence simulations was assigned as 763–795, based on the sequence numbering within the triple-helical region of type II collagen, instead of 963–995 as in the X-ray crystallographic structure.Molecular dynamics simulations
In order to explore the dynamic properties of MMP-1, we performed an extensive set of MD simulations for 300 ns (wild type MMP-1) and 100 ns for the modified linker systems (Table S1†) by using Gromacs 4.5.5 package21–23 with GROMOS96 43a1 (ref. 24) force field. The protonation states of His residues in the protein molecule were assigned based upon the optimal hydrogen bonding conformation performed in Gromacs using pdb2gmx. However H149, H164, H199, H203, and H209 were protonated at their delta and H177 at the epsilon position according to their local environment in the vicinity of Zn2+. The Zn2+ and Ca2+ ions were restrained in MD simulation according to their X-ray crystallographic structure distance using harmonic potential. In vacuo energy minimization was performed to remove steric clashes in the crystal structure first by using the steepest descent25 and then by using conjugate gradient26 until the maximum force was found smaller than 100 kJ mol−1 nm−1. The editconf command was used to define the dimension of the cubical box and the protein molecule was placed in the box. The periodic boundary conditions were then applied to treat all the parts of the system equally both at its interior and edges. The box size was set to ensure a distance of at least 1.0 nm between the protein and the box boundaries. The energy minimized protein structure was then solvated by using Single Point Charge (SPC)27 water model in the cubical simulation box by using Genbox command. The system was neutralized by adding Cl− to MMP-1 and the linker-modified enzymes (Table S1†). In order to relax the solvent molecule and remove constrains from the entire system, the energy minimization of the whole system was performed using first the steepest descent and then the conjugate gradient algorithm until the maximum force was found smaller than 100 kJ mol−1 nm−1. The energy minimized structure was then subjected to position restrain dynamics for 50 ps. The simulation was performed in NVT ensemble (constant number of particles, volume, and temperature)28 at constant temperature of 300 K with time step of 0.002 ps. The Berendsen temperature coupling and Parrinello–Rahman pressure coupling were used to keep the system at 300 K and 1 bar pressure during the simulation procedure. The productive MD was carried out using NVT ensemble and the initial velocities for MD simulation were drawn from Maxwell velocity distribution at 300 K. The MD was performed with an integration time step of 0.002 ps. The Particle Mesh Ewald (PME)29 method was used for electrostatic interactions with Coulomb cut off of 1.0 nm, Fourier spacing of 0.135 nm tolerance of 1 × 10−5 and an interpolation order of 4. The Lennard Jones potential was employed for the treatment of van der Waals interaction with cut off distance set to 1.4 nm. The LINCS algorithm30 was utilized to keep all the covalent bonds involving hydrogen atoms rigid.Analysis of molecular dynamics simulations
The analyses of the trajectories obtained from the simulations were performed using tools from the Gromacs software package. The Root Mean Square Deviation (RMSD) of Cα atoms of the protein with respect to minimized crystal structure, Root Mean Square Fluctuations (RMSF), electrostatic interactions, hydrogen bonding, Solvent Accessible Surface Area (SASA), and cluster analysis were performed. The visualization of MD trajectories and the structures were performed using VMD software.31 The Bio3D32 package in R was used to produce principal component (PC) analysis and domain cross correlation. Center of mass was calculated using Cα atoms of the CAT and HPX domains over simulation time using g_dist in Gromacs and also CALCOM33 to verify the results obtained from the g_dist in Gromacs.Dynamic cross correlation analysis
Correlated and collective atomistic motions play an important role in the functionality of dynamic biomolecular systems.34 The Cα atoms of the protein and THP were used to create a cross correlation Cij matrix using MD trajectory. The cross correlation between the ith and jth an atom were represented by a Cij matrix and was extracted by the following equation:| Cij = 〈Δri·Δrj〉/〈Δri2〉1/2〈Δrj2〉1/2 |
The Δri and Δrj are the displacement vectors corresponding to ith and jth atom and the angle brackets denote an ensemble average. The Cij matrix ranges from +1 to −1, where the positive value represented the correlated motion and the negative values represented the anticorrelated motion.
Results and discussion
Conformational dynamics of MMP-1·THP complex
The overall stability of the MMP-1·THP complex and changes associated within the internal structure and geometry of the individual CAT and HPX domains and linker region were assessed by computing the RMSD of the backbone Cα atoms with respect to the minimized X-ray crystallographic structure (PDB 4AUO). MMP-1·THP equilibrated after 10 ns with an average RMSD value of 4.7 Å (Fig. 2, S1A, and S1B, Table S2†). This RMSD is comparable to the RMSD of 4.2 Å of the collapsed (closed) form of MMP-1 in complex with the THP obtained from NMR studies.9 Three different runs of MMP-1 (Fig. S1B†) were performed in order to enhance the conformation sampling and avoid statistical errors, with an average value of 5.1 Å obtained (Table S3†). The multiple MD simulations were performed in order to evaluate the effect of the statistical noise on the quality of and to test the stability of the simulations. There is a discussion in the MD community about the impact of statistical errors on the quality of the MD simulations, and one of alternatives of single simulation is to run multiple MDs from the same starting structure with different initial velocities which allow evaluation of the quality of the single trajectories and to minimize the force-field related artifacts. The four simulations of MMP-1 show good consistence and convergence of the individual trajectories, as indicated by the RMSD plots (Fig. S1B†). Full length MMP-2 in a compact structure (PDB 1CK7) exhibited an average RMSD value of 4 Å during the first 70 ns of simulations performed by Diaz and co-workers.10 The MD simulation of MMP-2·fTHP-5 performed by Diaz and coworkers showed on average RMSD of ∼6 Å.35 The MMP-1 CAT domain equilibrated at ∼10 ns and was stable throughout the simulation with RMSD less than 2 Å (Fig. S1†). The HPX domain showed a drift at ∼60 ns with an average value of 2.6 Å and then stabilized for the entire length of the simulation (Fig. S1†). The linker region was also stable with an RMSD of 2 Å and showed equilibration at ∼10 ns (Fig. S1†). The linker region underwent limited structural deviation indicating that the inter-domain arrangement in MMP-1 in solvent is maintained close to the X-ray crystallographic structure throughout the simulation. The RMSD of the CAT and HPX domains and the linker of MMP-1 were comparable to the MD results from the simulation of the CAT domain (2.9 Å), HPX domain (2.4 Å), and linker region (2.1 Å) of MMP-2 complexed with THPs.36 The RMSD data suggested that the HPX domain shows greater structural deviation in comparison to the CAT domain. The simulations of MMP-2 also showed higher structural deviation of the HPX domain (1.7 Å) in comparison to CAT domain (1.1 Å) after 200 ns.11 The CAT and HPX domains in MMP-2·fTHP-5 showed RMSD values of ∼2 and ∼4 Å, respectively, and the HPX domain showed greater structural deviation in contrast to CAT domain.35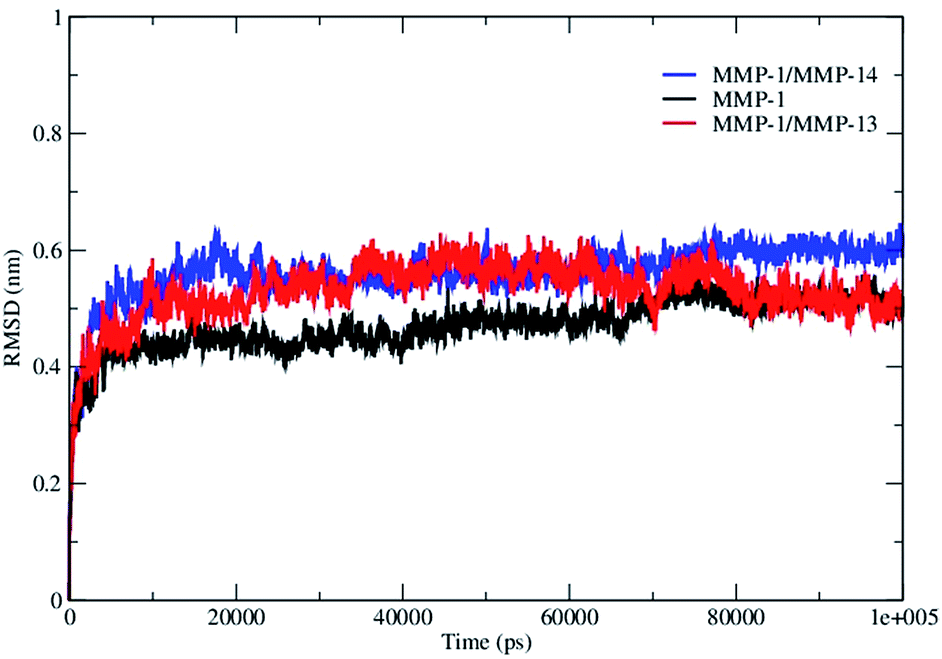 | ||
| Fig. 2 The RMSD of all Cα atoms of the MMP-1·THP complex. The RMSD of MMP-1 is compared with MMP-1 with the linker of MMP-14 (MMP-1/MMP-14) and MMP-1 with the linker of MMP-13 (MMP-1/MMP-13). | ||
The THP exhibited a high RMSD value (7 Å) relative to components of MMP-1 (Fig. S1†) indicating a significant structural change in comparison to the X-ray crystallographic structure. The THPs in the MMP-2 complex also exhibited a higher RMSD (on average 4.7 Å).36 In order to evaluate the compressing effect of the enzyme on the triple-helix dynamics, free THP in water was simulated. The average RMSD value of the THP in water was 22 Å with respect to 7 Å in the MMP-1·THP complex for 50 ns simulation (Fig. S1†), which implied a restraining effect of the enzyme on the THP and also highlighted the extent of conformational flexibility and dynamics of the THP in solution. Comparison of the averaged structures of the THP in water (based upon cluster analysis of the most populated cluster) and in the enzyme·substrate complex indicated considerable conformation changes (Fig. 3), although the triple-helical nature of the THP in water was maintained by consistent interchain hydrogen bonding. Enhanced conformational flexibility of THPs during MD has been reported previously37,38 as well the dynamic nature of the individual strands and their associated structures.49
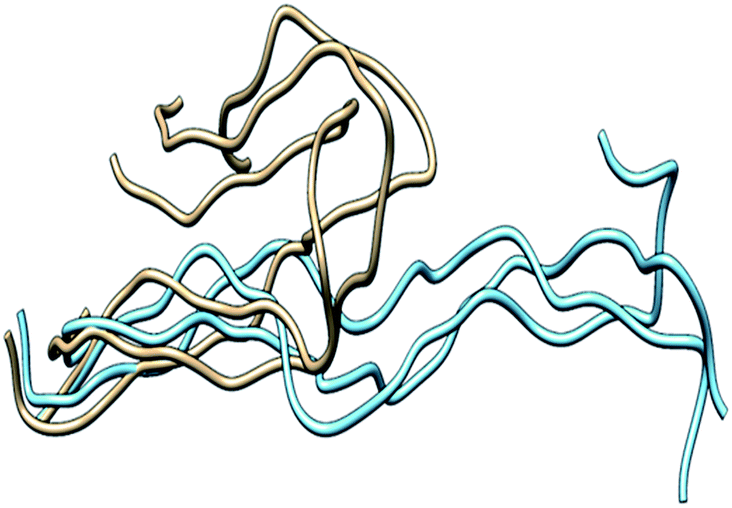 | ||
| Fig. 3 Superposition of the THP in water versus the THP in the MMP-1·THP complex. The structure of the most populated cluster of THP in water (brown) was aligned against the structure obtained from the most populated cluster of THP in the MMP-1·THP complex (cyan). | ||
The flexibility of MMP-1 and THP residues was assessed using RMSF analysis (Fig. 4A and S2A†). Average RMSF values were 1.14 Å for the CAT domain, 2.15 Å for the linker, 1.40 Å for the HPX domain, and 2.0 Å for the THP. The linker had fluctuations which peaked at 3.1 Å while the HPX domain displayed greater fluctuations relative to the CAT domain. The RMSF value of linker region in the compact conformation of MMP-2 was approximately 2.4 Å.39 The RMSF values of residues belonging to the HPX domain of MMP-2 also exhibited greater fluctuations in contrast to its CAT domain and linker.11,36 The loop region between parallel β-strands β2, β3, and β4 presented high fluctuations in the MMP-1 CAT domain. The αC of the CAT domain and the loop connecting the linker to β6 of the HPX domain showed increased flexibility. Increased fluctuations in the HPX domain was seen in the region encompassing residues of β17 and the loop between β17 and β18. The basal level fluctuations for MMP-1 were considered to be 1.4 Å (based upon the mean value), where 43% of the residues showed fluctuations greater than 1.4 Å and ∼56.9% of the residues showed fluctuations less than 1.4 Å (Table S2†).
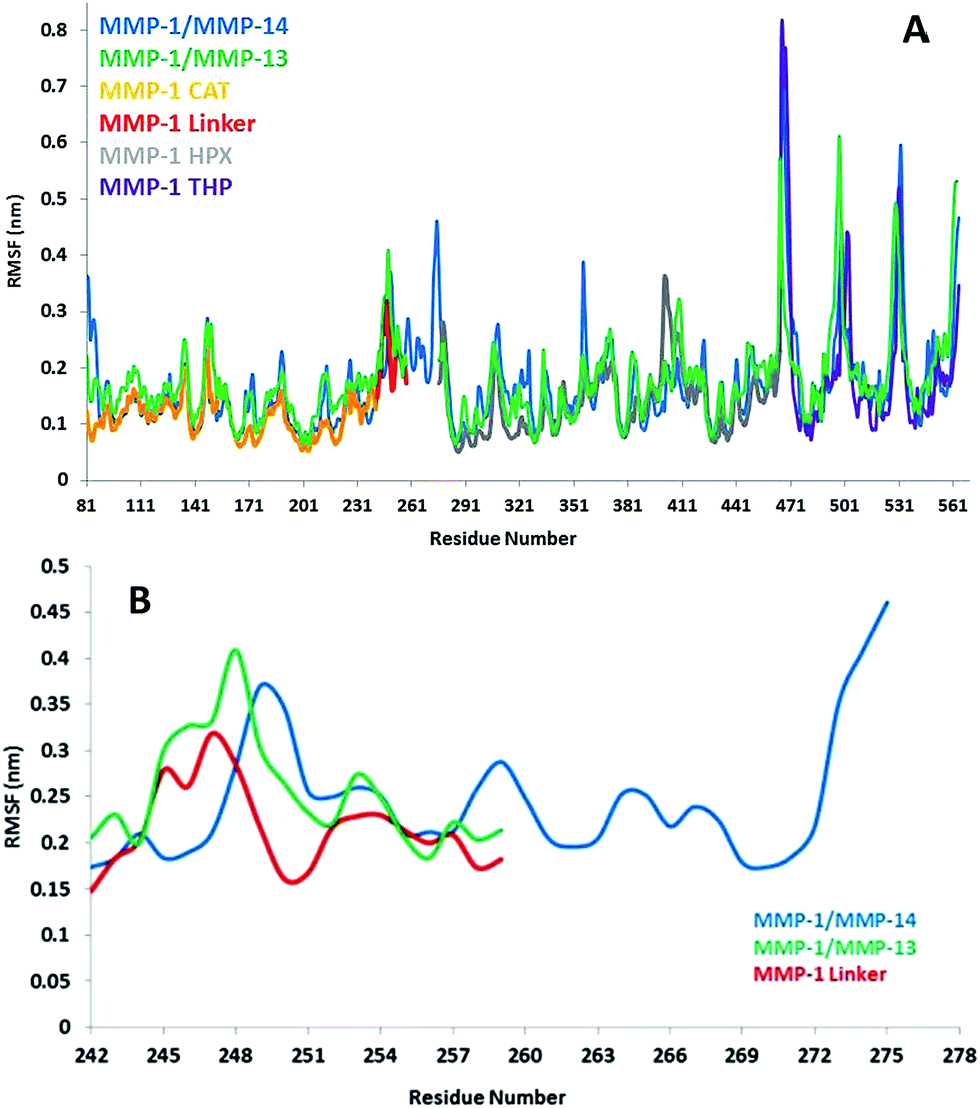 | ||
| Fig. 4 (A) RMSFs of MMP-1, MMP-1/MMP-14, and MMP-1/MMP-13 using Cα atoms. (B) RMSF of Cα atoms of the linker region of MMP-1 with respect to MMP-1/MMP-14 and MMP-1/MMP-13 averaged over 100 ns MD trajectory. The numbering of residues is according to the 4AUO X-ray crystallographic structure.8 | ||
The highest peak in the RMSF plot was from the N- and C-termini of the THP (Fig. 4A). The RMSF profile of the scissile bond (G775-L776) also showed high flexibility for the M and T strands with respect to the L strand (Fig. S2A†). In the MD simulations of MMP-2 the N- and C-termini of the THP bound to MMP-2 also exhibited high RMSF in the range of 8 to 16 Å, respectively.36
PC analysis can provide information on the overall dynamics of the THP in water. PC1 and PC2 accounted for ∼90% of the overall motion of the THP in solvent (Fig. S2B†). The maintenance of triple-helical structure and PC analysis of the THP in water shows good agreement with the fluorogenic THP simulation in aqueous solution where the first PC accounts for the twist motion of the triple-helix and the triple-helix is maintained throughout 50 ns trajectory.40 The projection of PC1 on the THP indicated bent or twist conformation as has been described by cluster analysis. The dynamics cross correlation analysis of the THP chains showed significant correlated motion through the 50 ns trajectory (Fig. S2C†). The extent of both positive and negative motion were in accordance with the maintenance of triple-helical structure during simulation.
The L strand of the THP approached closest to the active site in the simulation and exhibited the largest bending out of all of the THP strands (Fig. S3†). Experimental studies on MMP-1 (ref. 9, 14 and 15) suggested that the active site of MMP-1 could not accommodate the 15 Å diameter of the collagen triple-helix. Therefore, the sequential hydrolysis of single collagen strands occurs through the binding of the triple-helix to the HPX domain and allowing inter-domain conformational flexibility through the linker to facilitate the reorientation of CAT domain to position itself to perform catalysis. The reaction mechanism of collagen hydrolysis by MMPs requires the carbonyl group of the scissile peptide bond to coordinate with the catalytic Zn2+ in active site.39,41 The scissile bond between residues G775 and L776 of the L strand is located 8.8 Å from the catalytic Zn2+ in the CAT domain; therefore, the X-ray crystallographic structure of MMP-1 bound to the THP is unproductive due to absence of the scissile bond near the S1–S′1 subsites in the CAT domain. In the MD simulations, the bond distance decreased on average to 6.7 Å (Fig. S4†) but still does not represent the active complex.
Triple–helix interactions with the CAT and HPX domains
It has been demonstrated experimentally that specific residues present in blades 1 and 2 of the HPX domain interact with the collagen triple-helix.8,9,13,14 In the present simulation specific residues within blade 1 (residues 250–300) belonging to β6–β9 (Fig. 1B) interacted with the THP (Fig. S5†). New sets of interactions emerged between the L strand of the THP and the HPX domain. The backbone of L295 makes hydrogen bonds with the side chain of R780 with an average distance of 3.5 Å. The side chain of N296 participated in hydrogen bonding with the backbone of G781 and hydrophobic interaction with the aliphatic side chain of R780. The guanidino group of R780 had a close interaction with the carboxylic group of E294 (average distance of 4.5 Å) with a weak electrostatic interaction between the residues.The X-ray crystallographic structure revealed a hydrophobic cluster formed by V300 and F301 side chains with V783 of the L THP strand.8 This interaction was lost in the MD simulation and a new hydrophobic interaction between the side chains of P303 and I782 was established (Fig. S6†). A hydrogen bond also emerged between the side chain of Q335 and G784 (L strand) (Fig. S6†). R780 to G784 are located in the THP strands which are most bent during the simulation. R780 makes extensive interactions with the HPX domain and could be a reason why the THP is so bent during the simulation. The M strand of the THP made most of the interactions with the HPX domain in similar fashion to the X-ray crystallographic structure; nonetheless, new interactions emerged from the simulation studies, specifically involving T270 and N296 (Fig. S7†). R272 made hydrogen bonds with the backbone and side chain of O786 (M strand) and R789 (M strand), respectively, as described in the X-ray crystallographic structure. The aliphatic region of R272 was involved in formation of a hydrophobic cluster with the side chains of R789 and L785 of the M THP stand (Fig. S8†).
We previously demonstrated experimentally that the MMP-1 HPX domain binds to the THP with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry.9 This suggested that the HPX domain binds to more than one THP chain. The MMP-1 X-ray crystallographic and NMR structures8,9 showed the interaction of L and M strands of the THP with residues of blades 1 and 2 of the HPX domain. Our MD simulation also showed the existence of interactions between the HPX domain and two THP strands (L and M).
:1 stoichiometry.9 This suggested that the HPX domain binds to more than one THP chain. The MMP-1 X-ray crystallographic and NMR structures8,9 showed the interaction of L and M strands of the THP with residues of blades 1 and 2 of the HPX domain. Our MD simulation also showed the existence of interactions between the HPX domain and two THP strands (L and M).
The CAT domain mainly interacts with the L strand of the THP (Table S4†). The X-ray crystallographic structure indicated that the Y221 backbone formed a hydrogen bond with the side chain of Q779 (L strand); however, the simulation results revealed that the side chain of Q779 (L strand) moves away with an average distance of 5.5 Å from the backbone NH group of Y221 and makes a close interaction in the S1 subsite of the CAT domain (Fig. S9†). The MD studies confirm the presence of the Q779 side chain (L strand) in the S1 subsite with an average distance of 4.7 Å (Fig. S10†) further indicating the absence of the scissile bond near the active site. Our MD shows no significant interactions been made by a residue of the CAT domain with residues in vicinity of scissile bond, thus confirming the unproductive orientation in the X-ray crystallographic structure.
Flexibility and domain interactions in MMP-1 with linker regions from MMP-14 or MMP-13
The linker regions in MMPs have been proposed to play important roles for the mutual orientation of the domains and their interactions with collagen.9,14,15 In order to explore the effects of the nature of the MMP-1 linker region (both the length of the linker and its amino acid sequence) on modulating domain interactions, it was exchanged with the linker region from MMP-14 (which contains 35 residues compared with 17 residues in MMP-1) or MMP-13 (which consists of the same number of residues as the MMP-1 linker but with a different sequence).42 The trajectory of MMP-1 with in silico MMP-14 linker (MMP-1/MMP-14) showed that the system equilibrated at ∼20 ns (Fig. S11†). The average RMSD value of MMP-1/MMP-14 was 5.6 Å compared to 4.7 Å for MMP-1 (Fig. 2). Comparison of the RMSD of the CAT and HPX domains of MMP-1/MMP-14 with MMP-1 indicated that the CAT domain of MMP-1/MMP-14 showed slightly higher structural deviation in comparison to MMP-1 (Fig. S12†). The average RMSD values of the linker region are 5.6 Å for MMP-1/MMP-14 and 2.0 Å for MMP-1 (Fig. S13†) indicating greater structural deviation in the former case. These differences could have significant impact on interactions between the CAT and HPX domains and would affect the conformational dynamics of both domains.The basal level fluctuation for MMP-1 was considered to be 1.4 Å. In MMP-1/MMP-14 57% of the residues showed fluctuations greater than 1.4 Å (Table S2†) indicating an overall increase of 14% higher fluctuations in MMP-1/MMP-14 compared to the RMSF of MMP-1 (Fig. S14†). This increase could be attributed to the larger size of the MMP-14 linker (35 amino acids) compared to the 17 amino acid residue linker of MMP-1. The RMSFs for both domains of MMP-1/MMP-14 showed on average >10% overall increase in comparison to MMP-1 (Table S5†). The THP complexed to MMP-1/MMP-14 showed 20% increased fluctuations with respect to the THP complexed to MMP-1 (Table S5†). The overall effect of the MMP-14 linker in MMP-1 was an increase in the flexibility of the enzyme·substrate complex. Residues 171–180 encompassing the loop between β4 and β5 exhibited increased fluctuations in MMP-1/MMP-14. These residues are in vicinity of the structural Zn2+ in the CAT domain. Residues 188–191 encompassing the loop between β5 and αB also showed increased fluctuations in MMP-1/MMP-14. The adjacent αB helix contains H199, E200, and H203, which are involved in coordination with the catalytic Zn2+ in the CAT domain. The residues of the loop between α-helices αB and αC showed increased flexibility. This loop contains H209, which is coordinated to the catalytic Zn2+. There was significantly more fluctuation in the HPX domain compared to CAT domain of MMP-1/MMP-14 (Fig. S14†).
The linker region in MMP-13 is the same size as the linker in MMP-1, but with a different sequence.42 The RMSD profile of MMP-1 with the linker of MMP-13 (MMP-1/MMP-13) showed that the system equilibrated at ∼20 ns (Fig. 2) and indicated that MMP-1/MMP-13 had greater structural deviation with average value of 5.3 Å in comparison to MMP-1 (4.7 Å). The RMSD profile of the linker in MMP-1/MMP-13 showed a similar trend to the MMP-1 linker, but exhibited higher flexibility (Fig. S15†). The RMSF plot of MMP-1/MMP-13 showed overall increased fluctuations in comparison to MMP-1 (Fig. S16†). In MMP-1/MMP-13 68% of the residues exhibited fluctuations greater than 1.4 Å (Table S2†) indicating an overall increase of 25% higher fluctuations compared to the RMSF of MMP-1. The RMSF of the two domains of MMP-1/MMP-13, in contrast to MMP-1, showed that the CAT domain of MMP-1/MMP-13 contains on average 64% residues with RMSF > 1.4% with respect to 31.6% in MMP-1. The HPX domain of MMP-1/MMP-13 contained 58.5% of residues with fluctuations >1.4% as compared to 46.8% in MMP-1. Comparison of the RMSF of the linker region residues showed an increase in comparison to MMP-1 that is slightly higher than in the linker of MMP-1/MMP-14 (Fig. 4B). An increase in the flexibility of residues in the CAT domain of MMP-1/MMP-13 was seen in the loop connecting αB and αC. The αC is directly connected to the linker region of MMP-1/MMP-13.
In order to understand how the inter-domain distance between CAT and HPX domain changes as a function of time we computed the distance between centers of mass of the CAT and HPX domains. The average inter-domain distances for MMP-1, MMP-1/MMP-14, and MMP-1/MMP-13 were 37.8, 37.0, and 37.7 Å, respectively, and remained relatively stable throughout the simulation (Fig. S17†). The inter-domain distance of MMP-1/MMP-14 did show a decrease after 80 ns simulation (Fig. S17†). The center of mass between the CAT and HPX domain in MMP-2 also showed similar trends with an average distance of 39.6 Å and remained stable throughout the entire length of simulation.11,36 The average distance between the scissile bond of the THP L strand and the CAT domain (catalytic Zn2+) was 6.7 Å in MMP-1, 8.4 Å in MMP-1/MMP-14, and 8.3 Å in MMP-1/MMP-13 (Fig. S18 and S19†), illustrating that changing the linker does not lead to a more productive conformation, but rather the opposite.
Triple-helix interactions with MMP-1/MMP-14 and MMP-1/MMP-13
The average bending angle during the MD simulation for the THP leading strand was 115.8° in MMP-1/MMP-14 and 86.4° in MMP-1 (Fig. S3 and S20†). The bending angle was also increased in MMP-1/MMP-13 compared with MMP-1 (Fig. S21†), suggesting that the triple-helix is accommodated in different orientations in MMP-1/MMP-14 and MMP-1/MMP-13 in comparison to MMP-1.The backbone of Y221 in MMP-1 formed hydrogen bonds with the backbone of R780 of the L THP strand (Table S4†). However, in MMP-1/MMP-14 this interaction was not maintained during the simulation (Fig. S22†). The side chain of Q779 of the THP L strand no longer accessed the active site of MMP-1/MMP-14 and was on average 10.3 Å away, while in MMP-1 it was 4.7 Å from the active site. The backbone of S220 formed hydrogen bonds with the side chain of Q779 after 40 ns simulation in MMP-1 (Table S4†), while there were no hydrogen bonds formed between the S220 backbone and the side chain of Q779 in MMP-1/MMP-14 (Fig. S23†). R780 of the THP L strand interacts with the MMP-1 HPX domain, while all of the interactions of R780 seem to be very unstable in the MMP-1/MMP-14 complex throughout the entire length of the simulation (Fig. S24†). R272 of the HPX domain was extensively involved in hydrogen bonding with the L785, O786, and R789 residues of the THP M strand in MMP-1. In MMP-1/MMP-14 the side chain of R272 makes a stable interaction with the backbone of O786 for 40 ns and disappears as the simulation evolved over time (Fig. S25†).
The change in atomistic interactions in MMP-1/MMP-14 in comparison to MMP-1 pinpoints the role of the linker in modulating the interactions between the enzyme and substrate. The secondary structure of the linker region of MMP-1 appears relatively stable as a function of simulation time (Fig. S26†). However, the linker of MMP-1/MMP-14 showed changes in secondary structure during the simulation (Fig. S27†).
In MMP-1/MMP-13, the Q779 side chain of the THP L strand accessed the active site S1 pocket with an average distance of 3.3 Å compared to 4.7 Å in MMP-1. The backbone of Y221 and P219 in MMP-1/MMP-13 made a hydrogen bond with R780 of the THP L strand in a similar manner as in MMP-1. The side chain of Q167 also made hydrogen bonds with the O771 side chain of the L strand as it did in MMP-1 (Fig. S28†). Residues of the HPX domain such as N296, T270, and R272 showed a similar pattern of interaction as MMP-1/MMP-14 (Fig. S29†). These results indicated that overall the interactions of the CAT domain with the THP in MMP-1/MMP-13 is closer to MMP-1, while the HPX domain interactions of MMP-1/MMP-13 showed a pattern which is closer to MMP-1/MMP-14 than MMP-1. The structures obtained from the most populated clusters from cluster analysis of MMP-1, MMP-1/MMP-13, and MMP-1/MMP-14 showed significant differences in orientation and binding of the THP to both the CAT and HPX domains (Fig. 5 and 6). The linker clearly modulates important interactions in both the CAT and HPX domains.
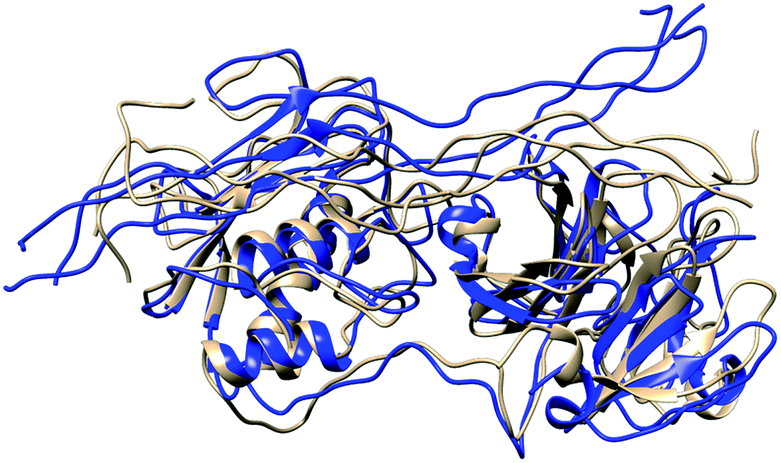 | ||
| Fig. 5 Structures obtained from the most populated cluster of MMP-1 (brown) and MMP-1/MMP-13 (blue). | ||
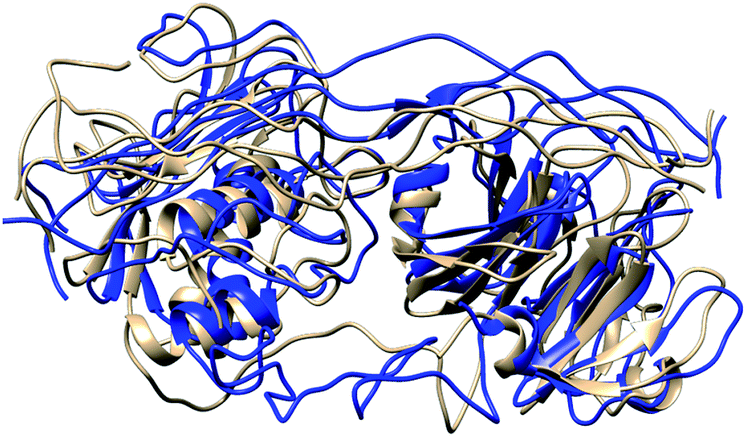 | ||
| Fig. 6 Structures obtained from the most populated cluster of MMP-1 (brown) and MMP-1/MMP-14 (blue). | ||
Correlated motions of the HPX and CAT domains: influence of the linker
The main aim of cross correlation analysis was to understand correlated motions between the MMP-1 HPX and CAT domains and to see how both domains are related to each other in the presence of triple-helices (Fig. 7A, cross-correlation map; Fig. 7B, elements from the molecular structure of MMP-1 involved in correlated and anti-correlated motions). Residues 105–115 of the CAT domain αA showed correlated motions with residues 175–195 of β5. The region 175 to 195 contains H164 and H177, which are coordinated to the structural Zn2+ and are located on a β-strand. The region from 182 to 194 belongs to loop which connects this β-sheet to an α-helix containing H199 and E200 which are coordinated to the catalytic Zn2+. The residues ranging from 140–150 of β3 and the loop joining β4 showed correlation towards residues 170–180 of β5 of the CAT domain.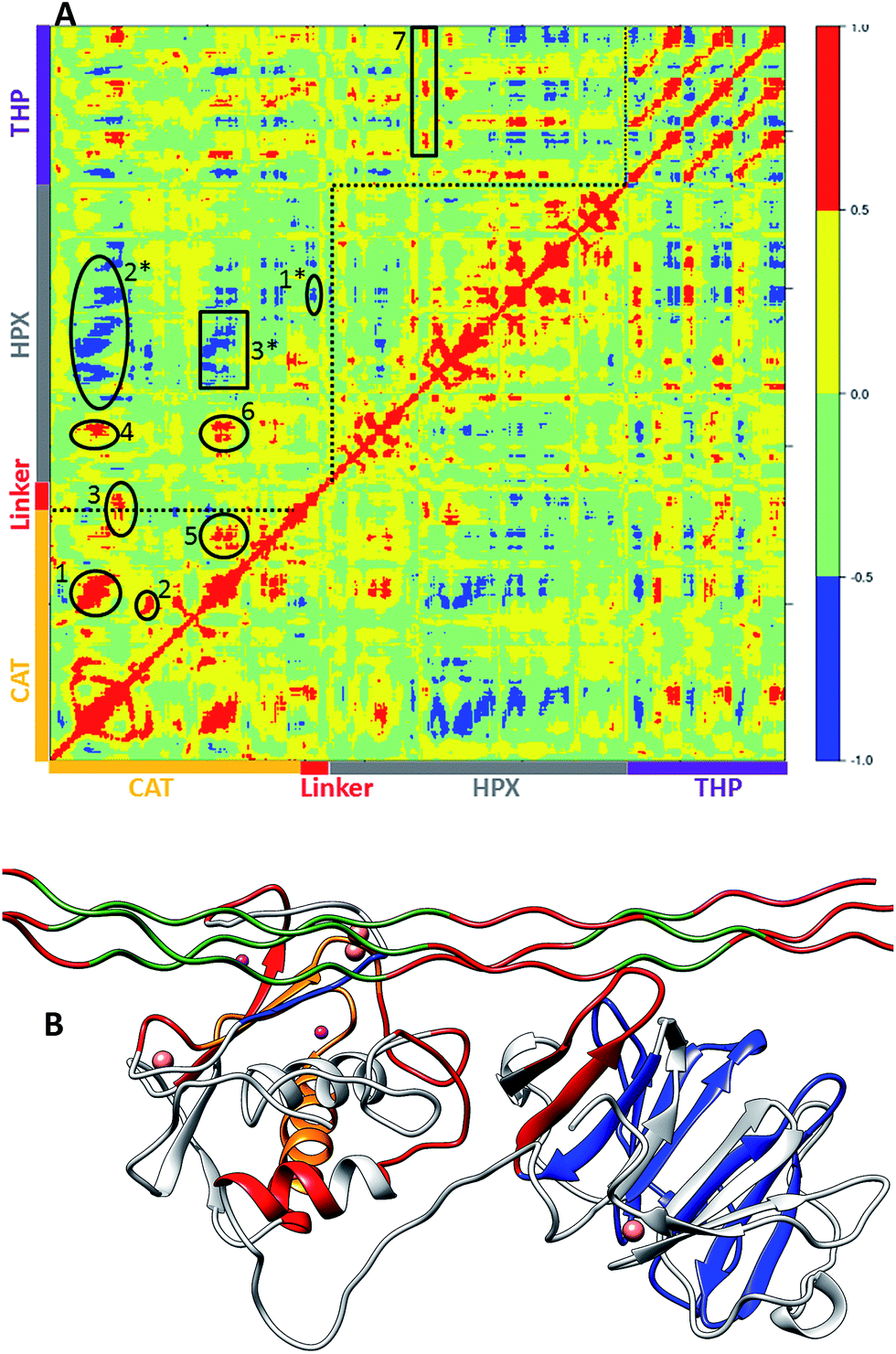 | ||
| Fig. 7 Dynamic cross correlation analysis of MMP-1·THP. (A) Intensities are indicated by the bar on the right. Numbering of correlations corresponds to specific interactions as follows: (1) the CAT domain residues 105–115 show correlation towards residues 175–195. (2) Residues 140–150 show correlation towards residues 170–180. (3) Residues 117–125 show correlation to residues 235–240. (4) CAT domain residues 105–115 show correlation towards HPX domain residues 287–294. (5) CAT domain residues 180–195 show correlation towards CAT domain residues 220–230. (6) Residues 180–195 show correlation towards HPX domain residues 280–290. Residues 180–195 also show correlation to THP residues 780–785. (7) HPX domain residues 315–320 are correlated to the ends of the THP strands. (1*) Residues 160–165 show negative correlation to HPX domain residues 275–280. (2*) CAT domain residues 100–120 show negative correlation towards HPX domain residues 310–380. (3*) CAT domain residues 175–185 show negative correlation towards HPX domain residues 330–350. (B) The correlated and anti-correlated motions between the residues of MMP-1·THP are projected on to the 3-D structure, where red and blue follows the scheme above and orange shows residues participating in both correlated and anti-correlated motion. The Zn2+ and Ca2+ are shown in purple and pink, respectively. The THP and MMP-1 are shown in green and grey, respectively. | ||
The β3, β4, and β5 are arranged in parallel conformation to each other in the X-ray crystallographic structure (Fig. 1). These β-sheets also surround the structural Zn2+ of the CAT domain. Residues 117–125 of αA showed correlation to residues 235–240 of αC, which is directly connected to the linker region. There was also correlation observed between residues 105–115 of αA of the CAT domain and residues 287–294 of the loop region between β8 and β9 of the HPX domain. Residues 180–195 of the loop region between β5 and αB of the CAT domain showed correlation towards residues 220–230 of the loop region between αB and αC of the CAT domain. The loop encompassing residues 220–230 is in very close proximity to the THP L strand. Residues 180–195 show correlation towards HPX domain residues 280–290. Residues 180–195 also show correlation to THP residues 780–785. This is the region of the THP showing local destabilization. HPX domain residues 315–320, belonging to the loop connecting two β-sheets together, are correlated to the ends of the THP strands. Overall, there was limited correlated motion seen between the HPX and CAT domains suggesting limited influence of the HPX domain on the conformational dynamics of the CAT domain via the linker.
Cross correlation analysis of MMP-1/MMP-14 and MMP-1/MMP-13 was subsequently performed (Fig. 8 and 9, cross-correlation maps; Fig. S30,† the correlated elements from the protein structures). The terminal residues of the linker region of MMP-1/MMP-14 showed correlation with the loop region of the C-terminus (residues 439–447) of the HPX domain. Residues 422–427 of β22 of the HPX domain also showed correlation towards the residues of the linker region. The residues in the loop connecting the C-terminus of the HPX domain showed positive correlation toward the linker region residues. Residues 409–413 of β20 showed correlation towards residues 262–267 located in vicinity of the linker region in the HPX domain. One important observation was the low degree of correlated motions observed between the THP and the CAT and HPX domains of MMP-1/MMP-14 in comparison to MMP-1. Interestingly, there was significant positive correlation between the residues of the linker region and the HPX domain in MMP-1/MMP-14. Overall, MMP-1 has more correlated and anti-correlated motion than MMP-1/MMP-14.
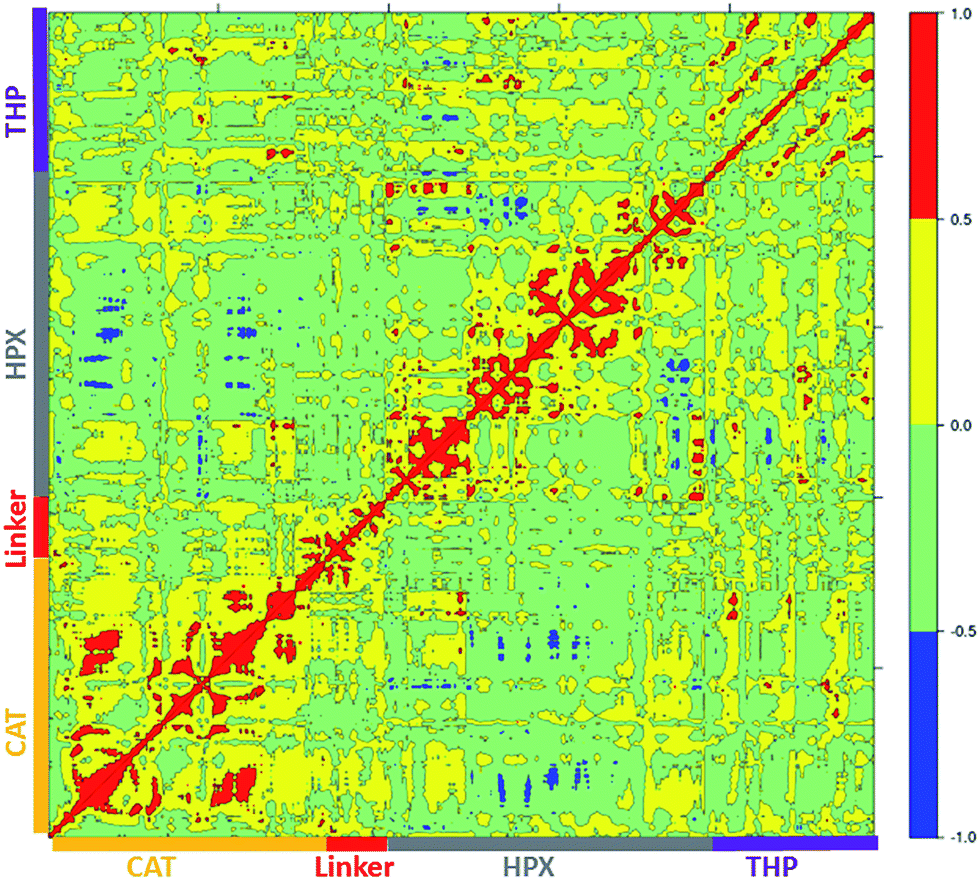 | ||
| Fig. 8 Dynamic cross correlation analysis of MMP-1/MMP-14·THP. Intensities are indicated by the bar on the right. | ||
 | ||
| Fig. 9 Dynamic cross correlation analysis of MMP-1/MMP-13·THP. Intensities are indicated by the bar on the right. | ||
The terminal residues of the MMP-1/MMP-13 linker region showed correlated motion towards the C-terminal residues (436–444) and β21 of the HPX domain. The residues of the loop region between β8–β9 (288–296) showed correlation towards the C-terminal residues (436–444) of the HPX domain. Residues of the linker region of MMP-1/MMP-13 and MMP-1/MMP-14 have positive correlation towards the HPX domain, which was not observed with the natural linker of MMP-1. The greater extent of positive and negative correlation in MMP-1/MMP-13 in contrast to MMP-1 indicated that even changes in the sequence of the linker produces significant differences in the motion of residues.
Conclusions
MD simulations of the X-ray crystallographic structure of MMP-1 (ref. 8) revealed that conformational changes exercise an important role and influence the interactions between the enzyme HPX and CAT domains and revealed the existence of extensive correlated motions. The interactions between the THP chains and the enzyme also have a flexible nature. Importantly, we confirm prior NMR experimental results9 that the X-ray crystallographic structure does not represent the functional enzyme·substrate (E·S) complex. Although we used this non-productive enzyme–substrate complex as a starting point for the MD simulations, as opposed to the productive complex reported from NMR spectroscopic studies, it is important to note that the X-ray crystallographic structure of MMP-1·THP is the only structure that defines the specific molecular interactions between the enzyme and substrate, as the NMR-derived structure is based on docking of residues. The MD simulation results are relevant to MMP-1 catalyzed collagenolysis, as the X-ray crystallographic and NMR structures of MMP-1·THP agree on the majority of THP interactions with the HPX domain and agree on the “closed” conformation of MMP-1 during collagenolysis. The significant point of departure in the two structures is the position of the THP with regards to the CAT domain, where the NMR-derived structure represents a productive complex while the X-ray crystallography-derived structure does not. The effects of these differences on MMP-1 dynamics and correlations can be explored in subsequent studies using MD simulations.The correlated motions observed here between the residues from the CAT domain and locally destabilized THP region would be important for the formation of the productive E·S complex. The correlated motions would also support a more favourable orientation of the enzyme electrostatic environment that would facilitate the catalyzed hydrolysis reaction in the active site (e.g., stabilizing the transition state). The simulations indicate the importance of implementing multiple structures from MD trajectories for reaction path calculations, as opposed to single structure based-calculations. The effect of the conformational flexibility of the E·S complex on the enzyme reaction path has already been demonstrated in number of mechanistic studies, such as cyclooxygenase-1.48
The linker plays a key role in modulating the interactions between the domains and formation of the active complex. The MD simulations with exchanged linker regions from MMP-14 and MMP-13 reveal the important role of the linker region to influence overall enzyme flexibility, the pattern of the correlated motions between the HPX and CAT domains, and the interactions between the triple-helical substrate and the enzyme. Prior experimental studies examined the effects of MMP linker exchange on collagenolysis. A chimeric MMP-8 whose linker region (17 residues) was replaced with the corresponding MMP-3 sequence (26 residues) lost activity towards collagen.43 In similar fashion, MMP-1/MMP-3 chimeras possessing the MMP-3 linker were not active towards collagen.44,45 The linker has been proposed to be critical for proper orientation of the CAT and HPX domains.9,46,47 The present study furthers the role of the linker, as it was found to facilitate correlative interactions between MMP domains and MMP domains and triple-helices. Overall, examination of MMP-1·THP demonstrates the potential of MD simulations to provide additional insight into experimentally-derived data.
Author contributions
W. S. carried out the MD simulations, analyzed and discussed the results, and wrote the manuscript. GF, C. C. and T. K.-C designed the study, analyzed and discussed the results, and wrote the manuscript.Acknowledgements
TK and CC acknowledge Marie Curie International Career Development Fellowships, NSCCS grants, and HEC-Biosim grants. WS thanks Northumbria University for a Ph.D. Studentship. GBF acknowledges support from the NIH (CA098799). The authors acknowledge Northumbria University Departmental HPC-Cluster “Pasteur”.Notes and references
- K. E. Kadler, C. Baldock, J. Bella and R. P. Boot-Handford, Collagens at a glance, J. Cell Sci., 2007, 120, 1955–1958 CrossRef CAS PubMed.
- M. Egeblad and Z. Werb, New functions for the matrix metalloproteinases in cancer progression, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- F. Song, K. Wisithphrom, J. Zhou and L. J. Windsor, Matrix metalloproteinase dependent and independent collagen degradation, Front. Biosci., 2006, 11, 3100–3120 CrossRef CAS.
- B. Fingleton, Matrix metalloproteinases as valid clinical targets, Curr. Pharm. Des., 2007, 13, 333–346 CrossRef CAS PubMed.
- S. Amar and G. B. Fields, Potential clinical implications of recent matrix metalloproteinase inhibitor design strategies, Expert Rev. Proteomics, 2015, 12, 445–447 CrossRef CAS PubMed.
- A. Page-McCaw, A. J. Ewald and Z. Werb, Matrix metalloproteinases and the regulation of tissue remodelling, Nat. Rev. Mol. Cell Biol., 2007, 8, 221–233 CrossRef CAS PubMed.
- G. B. Fields, Interstitial collagen catabolism, J. Biol. Chem., 2013, 288, 8785–8793 CrossRef CAS PubMed.
- S. W. Manka, F. Carafoli, R. Visse, D. Bihan, N. Raynal, R. W. Farndale, G. Murphy, J. J. Enghild, E. Hohenester and H. Nagase, Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12461–12466 CrossRef CAS PubMed.
- I. Bertini, M. Fragai, C. Luchinat, M. Melikian, M. Toccafondi, J. L. Lauer and G. B. Fields, Structural Basis for Matrix Metalloproteinase 1-Catalyzed Collagenolysis, J. Am. Chem. Soc., 2012, 134, 2100–2110 CrossRef CAS PubMed.
- N. Diaz, D. Suarez and H. Valdes, From the X-ray compact structure to the elongated form of the full-length MMP-2 enzyme in solution: a molecular dynamics study, J. Am. Chem. Soc., 2008, 130, 14070–14071 CrossRef CAS PubMed.
- N. Diaz and D. Suarez, Alternative interdomain configurations of the full-length MMP-2 enzyme explored by molecular dynamics simulations, J. Phys. Chem. B, 2012, 116, 2677–2686 CrossRef CAS PubMed.
- K. G. Lu and C. M. Stultz, Insight into the degradation of type-I collagen fibrils by MMP-8, J. Mol. Biol., 2013, 425, 1815–1825 CrossRef CAS PubMed.
- J. L. Lauer-Fields, M. J. Chalmers, S. A. Busby, D. Minond, P. R. Griffin and G. B. Fields, Identification of specific hemopexin-like domain residues that facilitate matrix metalloproteinase collagenolytic activity, J. Biol. Chem., 2009, 284, 24017–24024 CrossRef CAS PubMed.
- L. H. Arnold, L. E. Butt, S. H. Prior, C. M. Read, G. B. Fields and A. R. Pickford, The interface between catalytic and hemopexin domains in matrix metalloproteinase-1 conceals a collagen binding exosite, J. Biol. Chem., 2011, 286, 45073–45082 CrossRef CAS PubMed.
- L. Cerofolini, G. B. Fields, M. Fragai, C. F. Geraldes, C. Luchinat, G. Parigi, E. Ravera, D. I. Svergun and J. M. Teixeira, Examination of matrix metalloproteinase-1 in solution: a preference for the pre-collagenolysis state, J. Biol. Chem., 2013, 288, 30659–30671 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera – a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- A. Stivala, M. Wybrow, A. Wirth, J. C. Whisstock and P. J. Stuckey, Automatic generation of protein structure cartoons with Pro-origami, Bioinformatics, 2011, 27, 3315–3316 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- N. Guex and M. C. Peitsch, SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling, Electrophoresis, 1997, 18, 2714–2723 CrossRef CAS PubMed.
- A. Fiser and A. Sali, Modeller: generation and refinement of homology-based protein structure models, Methods Enzymol., 2003, 374, 461–491 CAS.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- H. J. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: A message-passing parallel molecular dynamics implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, GROMACS: fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- L. D. Schuler, X. Daura and W. F. Van Gunsteren, An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase, J. Comput. Chem., 2001, 22, 1205–1218 CrossRef CAS.
- R. Fletcher and M. J. Powell, A rapidly convergent descent method for minimization, Comput. J., 1963, 6, 163–168 CrossRef.
- M. Hestenes and E. Stiefel, Methods of Conjugate Gradients for Solving Linear Systems, J. Res. Natl. Bur. Stand., 1952, 49, 409–436 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces, ed. B. Pullman, Springer, Netherlands, 1981, vol. 14, ch. 21, pp. 331–342 Search PubMed.
- I. R. McDonald, NpT-ensemble Monte Carlo calculations for binary liquid mixtures, Mol. Phys., 1972, 23, 41–58 CrossRef CAS.
- P. P. Ewald, Die Berechnung optischer und elektrostatischer Gitterpotentiale, Ann. Phys., 1921, 369, 253–287 CrossRef.
- B. Hess, P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation, J. Chem. Theory Comput., 2007, 4, 116–122 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. J. Grant, A. P. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. Caves, Bio3d: an R package for the comparative analysis of protein structures, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed.
- G. Chelvanayagam, L. Knecht, T. Jenny, S. A. Benner and G. H. Gonnet, A combinatorial distance-constraint approach to predicting protein tertiary models from known secondary structure, Folding Des., 1998, 3, 149–160 CrossRef CAS PubMed.
- B. L. Kormos, A. M. Baranger and D. L. Beveridge, Do collective atomic fluctuations account for cooperative effects? Molecular dynamics studies of the U1A-RNA complex, J. Am. Chem. Soc., 2006, 128, 8992–8993 CrossRef CAS PubMed.
- N. Diaz and D. Suarez, Extensive simulations of the full-length matrix metalloproteinase-2 enzyme in a prereactive complex with a collagen triple-helical peptide, Biochemistry, 2015, 54, 1243–1258 CrossRef CAS PubMed.
- E. R. Azhagiya Singam, V. Rajapandian and V. Subramanian, Molecular dynamics simulation study on the interaction of collagen-like peptides with gelatinase-A (MMP-2), Biopolymers, 2014, 101, 779–794 CrossRef CAS PubMed.
- E. Suarez, N. Diaz and D. Suarez, Entropic control of the relative stability of triple-helical collagen peptide models, J. Phys. Chem. B, 2008, 112, 15248–15255 CrossRef CAS PubMed.
- T. E. Klein and C. C. Huang, Computational investigations of structural changes resulting from point mutations in a collagen-like peptide, Biopolymers, 1999, 49, 167–183 CrossRef CAS PubMed.
- N. Diaz and D. Suarez, Peptide hydrolysis catalyzed by matrix metalloproteinase 2: a computational study, J. Phys. Chem. B, 2008, 112, 8412–8424 CrossRef CAS PubMed.
- N. Diaz, D. Suarez and H. Valdes, Unraveling the molecular structure of the catalytic domain of matrix metalloproteinase-2 in complex with a triple-helical peptide by means of molecular dynamics simulations, Biochemistry, 2013, 52, 8556–8569 CrossRef CAS PubMed.
- N. Diaz, D. Suarez and E. Suarez, Kinetic and binding effects in peptide substrate selectivity of matrix metalloproteinase-2: molecular dynamics and QM/MM calculations, Proteins, 2010, 78, 1–11 CrossRef CAS PubMed.
- M. J. Stawikowski and G. B. Fields, Matrix Metalloproteinases: From Structure to Function, in Matrix Metalloproteinase Biology, ed. I. Sagi and J. P. Gaffney, John Wiley & Sons, New York, 2015, ch. 1, pp. 1–22 Search PubMed.
- T. Hirose, C. Patterson, T. Pourmotabbed, C. L. Mainardi and K. A. Hasty, Structure–function relationship of human neutrophil collagenase: identification of regions responsible for substrate specificity and general proteinase activity, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2569–2573 CrossRef CAS.
- G. Murphy, J. A. Allan, F. Willenbrock, M. I. Cockett, J. P. O'Connell and A. J. Docherty, The role of the C-terminal domain in collagenase and stromelysin specificity, J. Biol. Chem., 1992, 267, 9612–9618 CAS.
- L. Chung, K. Shimokawa, D. Dinakarpandian, F. Grams, G. B. Fields and H. Nagase, Identification of the (183)RWTNNFREY(191) region as a critical segment of matrix metalloproteinase 1 for the expression of collagenolytic activity, J. Biol. Chem., 2000, 275, 29610–29617 CrossRef CAS PubMed.
- H. Tsukada and T. Pourmotabbed, Unexpected crucial role of residue 272 in substrate specificity of fibroblast collagenase, J. Biol. Chem., 2002, 277, 27378–27384 CrossRef CAS PubMed.
- G. F. Fasciglione, M. Gioia, H. Tsukada, J. Liang, R. Iundusi, U. Tarantino, M. Coletta, T. Pourmotabbed and S. Marini, The collagenolytic action of MMP-1 is regulated by the interaction between the catalytic domain and the hinge region, J. Biol. Inorg. Chem., 2012, 17, 663–672 CrossRef CAS PubMed.
- C. Christov, A. Lodola, T. Karabencheva, S. Wan, P. Coveney and A. Mulholland, Conformational Effects on the pro-S Hydrogen Abstraction Reaction in Cyclooxygenase-1: An Integrated QM/MM and MD Study, Biophys. J., 2013, 104, L5–L7 CrossRef CAS PubMed.
- I. Fu, D. Case and J. Baum, Dynamic Water-Mediated Hydrogen Bonding in a Collagen Model Peptide, Biochemistry, 2015, 54, 6029–6037 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03033e |
| This journal is © The Royal Society of Chemistry 2016 |