
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chitin-acetate/DMSO as a supramolecular green CO2-phile†
Ala'a F. Eftaihaa,
Fatima Alsoubania,
Khaleel I. Assafb,
Werner M. Naub,
Carsten Trollc and
Abdussalam K. Qaroush*cd
aDepartment of Chemistry, The Hashemite University, P.O. Box 150459, Zarqa 13115, Jordan
bDepartment of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany
cWACKER-Lehrstuhl für Makromolekulare Chemie, Technische Universität München, Lichtenbergstraße 4, 85747 Garching bei München, Germany. E-mail: abdussalam.qaroush@tum.de
dThe Jordanian Pharmaceutical Manufacturing Company (JPM), P.O. Box 94, Naour 11710, Jordan
First published on 18th February 2016
Abstract
The supramolecular chemisorption of CO2 by the oligomeric chitin-acetate (CA) in DMSO as a green solvent offers a novel eco-friendly approach for CO2 capture. Since the amino groups in the sorbent material are blocked either by protonation or acetylation, the multi-armed hydroxyl based oligosaccharide captures CO2 through the formation of an organic carbonate species as confirmed by 13C NMR, in situ ATR-FTIR spectroscopy and conductivity. DFT calculations verified the formation of the CA-CO2 adduct, in which the organic carbonate group is stabilized through supramolecular ionic interaction and hydrogen bonding with the neighboring ammonium ion and hydroxyl functional group along the oligomer backbone. The use of other polar aprotic solvents (N,N-dimethylformamide (DMF), acetonitrile, and acetone) was not successful due to solubility issues.
Introduction
Since the early days of the twentieth century, the use of monoethanolamine (MEA) as a scrubbing agent for CO2 capturing has become a mature technology1 with known drawbacks upon regeneration, viz., cross-linking, evaporation, and limited recoverability. Several alternatives have been evaluated, ranging from solid sorbents (porous, non-porous) to different liquid-based candidates (chemi- or physisorption).2 Most materials act as nitrogen-bearing donors rather than oxygen-based sorbents due to the greater basicity of the former that can react chemically with CO2 following carbamate formation.3 From a practical industrial point of view, any competitive technology needs to fulfil several critical parameters in terms of cost, eco-friendliness, recoverability, working temperature, efficiency of sorption and minimizing the use of sacrificial bases. One approach to increase the efficiency is the use of macromolecular, multi-reactive, and ideally bio-renewable materials to improve the stoichiometry of CO2 capture and reduce costs.4 Chitin and its hydrolyzed form, viz., chitosan, are potential candidates to replace MEA-based sorbents for CO2 capturing. What makes them appropriate as sorbents are their commercial availability, sustainability, mechanical strength and much more importantly is their nitrogen content. Solubility in common organic solvents and gelation are critical issues that hinder their mass use. Therefore, efforts are gathered to explore their CO2 capturing ability in ionic liquids (ILs).5,6Away from ILs, we directed our focus towards the use of oligosaccharide rather than their corresponding parent polymers. A careful compromise between solubility, concentration, and prevention of gelation were taken into consideration by tailoring the degree of deacetylation (DDA). As a continuation of our research on green sorbents7 for the capture of CO2, we have discovered a simple method which utilizes chitin-acetate (CA, Scheme 1) as a scrubbing agent in green solvent, namely, DMSO, by-passing the use of ILs8–10 and/or superbases.11
 | ||
| Scheme 1 Structural representation for CA as deduced by nuclear magnetic resonance, elemental analysis and potentiometric titrations. | ||
Results and discussion
Two physical parameters regarding chitin and chitosan, viz., solubility and gelation (vide supra) are taken into consideration as a proper choice of a scrubbing material with reduced viscosity.12,13 In fact, MEA solutions are always prepared with concentrations that are 20–40% (v/v) to diminish their corrosive character and increased viscosities upon capturing CO2. The search for scrubbing agents that can overcome such barriers, would be of great importance for both industrial and academic sectors. For the requested task, we were biased towards the use of low molecular weight chitin derivative as a green sorbent.Fig. 1 shows partial 13C NMR spectra before and after bubbling of CO2. A peak formed at 157.4 ppm indicates the presence of carbonate11,14 (no carbamate is formed as evidenced by 13C NMR).15 Precedent for chemisorption of CO2 through carbonate formation has been obtained by Stoddart and coworkers14 for cyclodextrins within extended metal-organic frameworks (MOFs). Herein, the choice of solvent proved to be critical. In particular, no reaction took place in aqueous solution. Presumably, the formation of the carbonate is facilitated in a polar aprotic solvent for kinetic (nucleophilicity) reasons. Herein, DMSO was used as a model solvent. The formation of carbonate at C-6 could be directly followed through the induced shift of the C-6 peaks at 60 ppm (13C NMR, Fig. 1), in which a clear splitting was observed after the CO2 bubbling (there was no shift for C-2 before and after bubbling which confirms the absence of carbamates. C-2 peaks were 57.4 and 57.3 ppm, respectively).
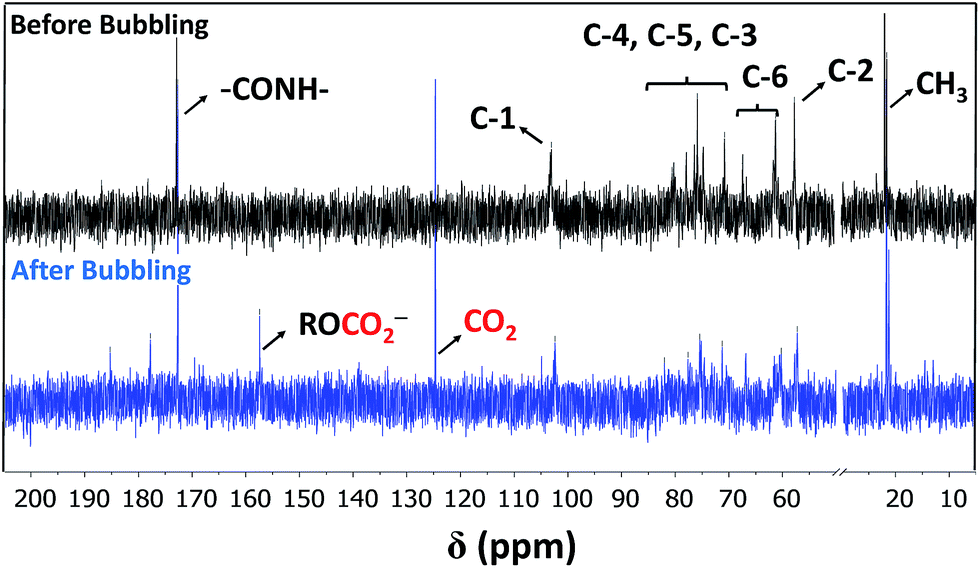 | ||
| Fig. 1 Partial 13C NMR spectra of CA dissolved in DMSO-d6 obtained before (black) and after (blue) bubbling of CO2. | ||
To differentiate between carbamates and carbonates, in situ ATR-FTIR measurements were carried out in the presence of CO2 in DMSO as a green solvent.16 Fig. 2 supports the assumption of chemisorption via two major peak changes. On one hand, the appearance of a new peak assigned to 1555 cm−1 that can be ascribed to the vibrational mode of carbonate.17 On the other hand, the presumed formation of carbamates (if any) should be accompanied with the emergence of a newly formed peak centered at ca. 1690 cm−1 (absent in our case).18 The peak centered at 1610 cm−1 is assigned to a bending mode of amine, together with the stretching mode of amide (I) at 1675 cm−1 that is overshadowed upon capturing of CO2.19 Further, the red shift in the (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) peak of acetate anion centered at 1720 cm−1 confirms the presumed interaction/metathesis of ammonium-acetate into ammonium-carbonate upon chemisorption of CO2.18
O) peak of acetate anion centered at 1720 cm−1 confirms the presumed interaction/metathesis of ammonium-acetate into ammonium-carbonate upon chemisorption of CO2.18
 | ||
| Fig. 2 Partial in situ IR spectra of CA (10.0% (w/v)) dissolved in DMSO obtained before (black) and after (blue) bubbling of CO2 ranging within (1500–1800) cm−1. | ||
A supramolecular stabilization of the organic carbonate species by ionic and hydrogen-bonding interactions of carbonate with neighbouring groups is presumed to be responsible for the observation that the formation of carbonates (–OH nucleophilic attack) was favoured over that of carbamates (amine nucleophilic attack). 13C NMR indicated that upon capturing CO2, the addition of water to the CA/carbonate adduct showed that the system is a thermodynamically stable due to the persistence of the organic carbonate peak. In addition, a newly formed peak at about 166.2 ppm indicated the formation of HCO3− because of interaction between physisorbed CO2 with water.
To prove the ion-pair formation in CA/CO2 adduct, the electrical conductivity of CA dissolved in DMSO was measured as a function of CO2 bubbling time (Fig. 3). The dropdown in conductivity is explained by the formation of neutral ionic aggregates rather than the presence of individually solvated ions (ammonium and acetate).20 Doubling of the polymer concentration resulted in an increased conductivity keeping up the same trend. This is considered as another proof of concept for the chemisorption as reported by Mu and co-workers.6
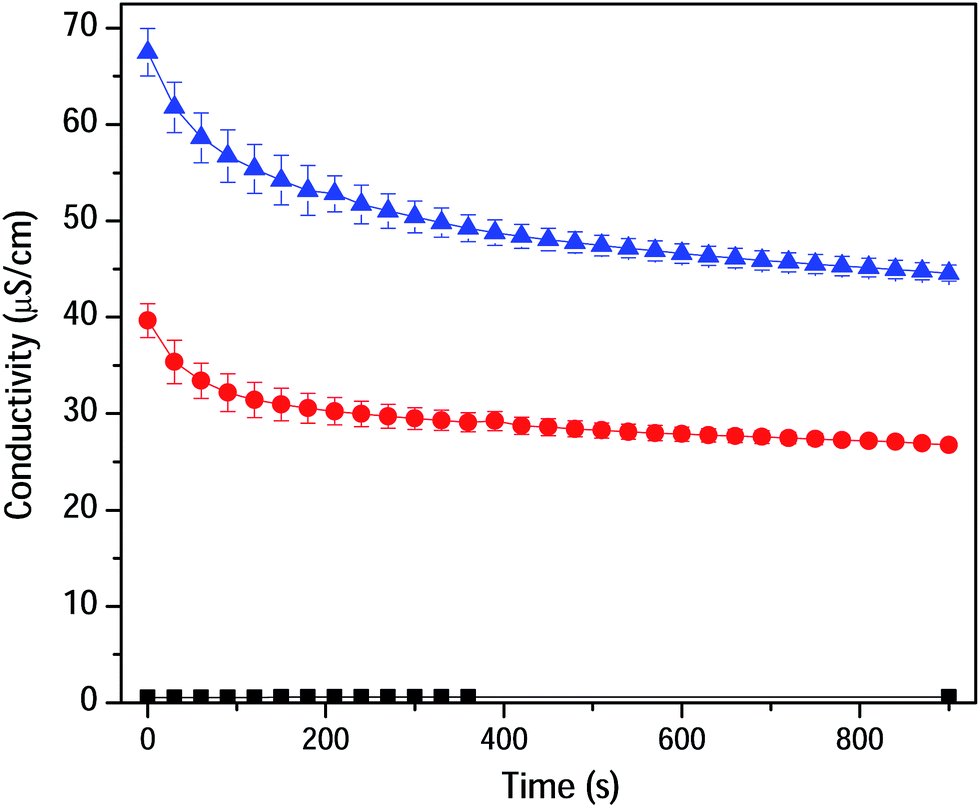 | ||
| Fig. 3 Conductivity of CA dissolved in DMSO as a function of CO2 bubbling time. DMSO and CA solutions (0.33 and 0.67 (w/v)%) are shown in black, red and blue traces, respectively. | ||
This notion was supported by DFT-calculated structures of the presumed carbonates in a trimer model, which assumed a comparable alternating conformation as in the biopolymer.21 The obtained geometry afforded close interaction distances between carbonate-ammonium ions (1.52 Å) and carbonate-hydroxyl groups (1.66 Å, Fig. 4).
 | ||
| Fig. 4 DFT-optimized (B3LYP/6-31G*) geometry (gas phase) for a glucosammonium trimer as a model compound. | ||
The results obtained from the above-mentioned analyses (NMR, in situ IR and DFT calculations) are described in the reaction shown in Scheme 2. The usage of DMSO would activate the hydroxyl group at C-6 of the amino pyranose ring toward nucleophilic attack, which result in chemisorption of CO2. The formed organic carbonate is stabilized via ionic interaction and hydrogen bonding along the oligomer backbone. To our knowledge, activation of alcohols by DMSO is a first of its kind. We tried to carry out the bubbling experiment in other polar aprotic solvent such as acetonitrile, acetone and N,N-dimethylformamide (DMF). Unfortunately, the insolubility of CA (in the former two solvents) or the limited solubility (in DMF) prohibited further investigation of carbonate formation (see Fig. S1, (ESI†)). This implies that solubility, dielectric constant and H-bond accepting character should be taken in consideration to carry out such reaction.
 | ||
| Scheme 2 The reaction of CA with CO2 in the presence of DMSO as a solvent; R = NH3+ or NHCOCH3. | ||
To measure the amount of sorbed CO2 by CA in DMSO volumetrically, pressurized vessels controlled with digital manometers were used. An arbitrary pressure was set at 4.0 bars to make sure it would remain in excess to achieve the equilibrated sorption capacities. The following conditions were adopted, namely 10.0% (w/v sorbent), 25.0 °C, and 4.0 bar CO2. Gas sorption was measured relative to neat DMSO, which is known to absorb one bar of CO2 at the same conditions, and a value of 3.63 mmolCO2 gsorbent−1 was determined for CA. This value is competitive to other scrubbing agents,2 which renders CA an interesting alternative from ecological and economic points of view. CO2 adsorption isotherm experiments at various pressure and temperature will be reported in an upcoming study to compare between the volumetric and gravimetric methods.
The reversibility of the process depends mainly on perturbing the supramolecular stabilized adduct. We tried several parameters e.g. pH change, sonication, thermal/electrical stimuli. Bubbles of CO2 were evolved upon applying the stress (vide supra). Therefore, the material is recyclable as an effective CO2 sorbent.
Experimental
Materials and methods
Unless otherwise stated, all chemicals were used without further purification. Chitin-acetate (CA) was made by G.T.C. Bio Corporation, Qingdao, China. For experimental manipulations, CA was dried in a vacuum oven at 50 °C, overnight. Number average molecular weight (Mn) was determined to be 7 kDa, by multiangle light scattering (MALS) analysis using a Wyatt Dawn Heleos II in combination with a Wyatt Optilab rEX as concentration Source (Wyatt, USA). Based on Mn value, CA comprised of 15 units. CO2 (99.95%, food grade) was purchased from advanced technical gases, Amman, Jordan. Dimethyl sulfoxide and DMSO-d6 were purchased from M-TEDIA and Sigma-Aldrich, respectively. Solution 13C-NMR spectra were collected at room temperature using AVANCE-III 400 MHz FT-NMR spectrometer NanoBay (Bruker, Switzerland) in either DMSO-d6 or D2O. In situ IR measurements were carried out using a MMIR45m RB04-50 (Mettler-Toledo, Switzerland) with an MCT Detector, with silicon windows probe connected via pressure vessel; sampling 3500 to 650 cm−1 at 8 wavenumber resolution; scan option: 64; gain: 1×. Conductivity measurements were carried out using 712 Conductormeter (Metrohm, Switzerland). Calculations were performed within Gaussian 09. The full optimization was performed using DFT method (B3LYP/6-31G*). Minima were characterized by the absence of imaginary frequencies.Conclusions
In conclusion, we propose a new method to absorb atmospheric CO2 (without the use of stoichiometric amounts of the superbases or ionic liquids) in the presence of DMSO as a green solvent. Whether the CO2 chemisorption by carbonate formation is favored for kinetic reasons (higher nucleophilicity in polar aprotic solvents) or thermodynamic reasons (supramolecular stabilization of the carbonate by interaction with neighboring groups) requires further investigations. Regardless of the mechanistic details, the formation of stable adducts in the capture of CO2 by green sorbents is of considerable practical interest. The commercial availability of this oligosaccharide and cheap cost for its processing are additional assets. The use of other polar aprotic solvents (N,N-dimethylformamide (DMF), acetonitrile, and acetone) was not successful due to solubility issues.Acknowledgements
A. F. E. and A. K. Q. thank Dr Adnan A. Badwan, the CEO of the Jordanian Pharmaceutical Manufacturing Company (JPM), Naor, Jordan, for the generous gift of CA. Moreover, thanks are paid to Prof. Dr Adnan S. Abu-Surrah and Prof. Dr Hamzeh M. Abdel-Halim for providing access to their laboratory, HU, Zarqa, Jordan.References
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
- X. Sun, C. Huang, Z. Xue and T. Mu, Energy Fuels, 2015, 29, 1923–1930 CrossRef CAS.
- A. K. Qaroush, D. A. Castillo-Molina, C. Troll, M. A. Abu-Daabes, H. M. Alsyouri, A. S. Abu-Surrah and B. Rieger, ChemSusChem, 2015, 8, 1618–1626 CrossRef CAS PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- P. S. Barber, C. S. Griggs, G. Gurau, Z. Liu, S. Li, Z. Li, X. Lu, S. Zhang and R. D. Rogers, Angew. Chem., Int. Ed., 2013, 52, 12350–12353 CrossRef CAS PubMed.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS PubMed.
- K. Kurita, Mar. Biotechnol., 2006, 8, 203–226 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed.
- F. Barzagli, F. Mani and M. Peruzzini, Energy Environ. Sci., 2009, 2, 322–330 CAS.
- US Environmental and Protection Agency, Dimethyl Sulfoxide Producers Association, Leesburg, VA, 2003 Search PubMed.
- S.-W. Song and S.-W. Baek, Electrochim. Acta, 2009, 54, 1312–1318 CrossRef CAS.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- D. Williams and I. Fleming, Spectroscopic methods in organic chemistry, McGraw-Hill, UK, 4th edn, 1989, Revised Search PubMed.
- T. Köddermann, S. Klembt, D. Klasen, D. Paschek, U. Kragl and R. Ludwig, ChemPhysChem, 2012, 13, 1748–1752 CrossRef PubMed.
- K. H. Gardner and J. Blackwell, Biopolymers, 1975, 14, 1581–1595 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03022j |
| This journal is © The Royal Society of Chemistry 2016 |