
Evolution of morphologies of a PE-b-PEO block copolymer in an epoxy solvent induced by polymerization followed by crystallization-driven self-assembly of PE blocks during cooling
Abstract
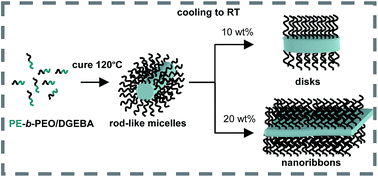
Polymerization-induced nanostructuration combined with crystallization-driven self-assembly was used to generate complex nanostructures in an epoxy network. A PE-b-PEO block copolymer (Mn = 1400; 50 wt% PEO), was dispersed in diglycidylether of bisphenol A (DGEBA) and homopolymerization initiated by a tertiary amine was carried out at 120 °C (above the melting temperature of PE). The plasticization produced by the miscible PEO blocks decreased the Tg of the cured matrix to values located below the crystallization temperature of PE. Therefore, crystallization-driven self-assembly of PE blocks took place during the cooling step through the rubbery region of the epoxy network. Depending on the initial amount of PE-b-PEO dispersed in DGEBA, a variety of nanostructures could be generated, such as a dispersion of disk-like micelles (6.7 nm in thickness), a concentrated dispersion of short nanoribbons (50–200 nm in length and 6.7 nm in thickness), partially stacked and oriented in space, and complex spherulitic structures composed of large stacked nanoribbons. The thickness of micellar objects was close to the theoretical value of fully extended PE chains of the block copolymer. IR spectroscopy confirmed the all-trans conformation of PE chains. Therefore, crystals were formed by interdigitated PE chains, with PEO blocks tethered at both planar interfaces in an alternating way. The way in which these complex nanostructures affect the fracture resistance or functional properties (such as shape memory) of the resulting epoxy networks has yet to be analyzed.
 Please wait while we load your content...
Please wait while we load your content...

