
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of the aromatic moiety in α- and β-arylalanines on their biotransformation with phenylalanine 2,3-aminomutase from Pantoea agglomerans†‡
Andrea
Varga
a,
Gergely
Bánóczi
b,
Botond
Nagy
a,
László Csaba
Bencze
a,
Monica Ioana
Toşa
a,
Ákos
Gellért
c,
Florin Dan
Irimie
a,
János
Rétey
d,
László
Poppe
*be and
Csaba
Paizs
*a
aBiocatalysis and Biotransformation Research Group, Babeş-Bolyai University of Cluj-Napoca, Arany János str. 11, RO-400028 Cluj-Napoca, Romania. E-mail: paizs@chem.ubbcluj.ro
bDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, Műegyetem rkp. 3, H-1111 Budapest, Hungary. E-mail: poppe@mail.bme.hu
cAgricultural Institute, Centre of Agricultural Research, Hungarian Academy of Sciences, Brunszvik u. 2, H-2462 Martonvásár, Hungary
dInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Richard-Willstätter-Allee, D-76128 Karlsruhe, Germany
eSynBiocat Ltd, Lázár deák u 4/1, H-1173 Budapest, Hungary
First published on 7th June 2016
Abstract
In this study enantiomer selective isomerization of various racemic α- and β-arylalanines catalysed by phenylalanine 2,3-aminomutase from Pantoea agglomerans (PaPAM) was investigated. Both α- and β-arylalanines were accepted as substrates when the aryl moiety was relatively small, like phenyl, 2-, 3-, 4-fluorophenyl or thiophen-2-yl. While 2-substituted α-phenylalanines bearing bulky electron withdrawing substituents did not react, the corresponding substituted β-aryl analogues were converted rapidly. Conversion of 3- and 4-substituted α-arylalanines happened smoothly, while conversion of the corresponding β-arylalanines was poor or non-existent. In the range of pH 7–9 there was no significant influence on the conversion of racemic α- or β-(thiophen-2-yl)alanines, whereas increasing the concentration of ammonia (ammonium carbonate from 50 to 1000 mM) inhibited the isomerization progressively and decreased the amount of the by-product (i.e. (E)-3-(thiophen-2-yl)acrylic acid was detected). In all cases, the high ee values of the products indicated excellent enantiomer selectivity and stereospecificity of the isomerization except for (S)-2-nitro-α-phenylalanine (ee 92%) from the β-isomer. Substituent effects were rationalized by computational modelling revealing that one of the main factors controlling biocatalytic activity was the energy difference between the covalent regioisomeric enzyme–substrate complexes.
Introduction
Nowadays there is an ever increasing demand for optically pure β-amino acids mainly by the pharmaceutical industry and peptide syntheses.1 In particular, the biological characteristics of the β-amino acids, along with their use as precursors of various heterocycles and as chiral auxiliaries in enantioselective syntheses have aroused lively interest in their chemistry. This induced rapid development of synthetic procedures for the preparation of enantiopure β-amino acids and their congeners.2 Until recently most of the biocatalytic approaches for them relied on kinetic resolution with hydrolytic enzymes, such as lipases,2 acylases3 and hydantoinases.4An attractive method for the synthesis of enantiopure, non-natural β-amino acids is based on the use of phenylalanine 2,3-aminomutase (PAM). According to their stereochemical preference there are two kinds of phenylalanine 2,3-amino-mutases (PAMs), the one of plant origin (EC 5.4.3.10) is producing (R)-β-phenylalanine,5 while the other one is of bacterial origin (EC 5.4.3.11) and is leading to (S)-β-phenyl-alanine.6,7 Both are members of the class I lyase-like family also including tyrosine 2,3-aminomutase (TAM, EC 5.4.3.6),8 phenylalanine ammonia-lyase (PAL, EC 4.3.1.24 and EC 4.3.1.25),9 tyrosine ammonia-lyase (TAL, EC 4.3.1.23 and EC 4.3.1.25),10 and histidine ammonia-lyase (HAL, EC 4.3.1.3).11 All of them utilize the same protein-derived prosthetic group, 3,5-dihydro-5-methylidene-4H-imidazol-4-one (MIO) (Fig. 1A), formed autocatalytically in the active site from an XSG motif which is typically Ala–Ser–Gly.11 Less frequently MIO could be formed from a Thr–Ser–Gly (in PaPAM and in SmPAM), a Ser–Ser–Gly (in HAL from Fusobacterium nucleatum) or a Cys–Ser–Gly (in HAL from Streptomyces griseus) motif as well.12
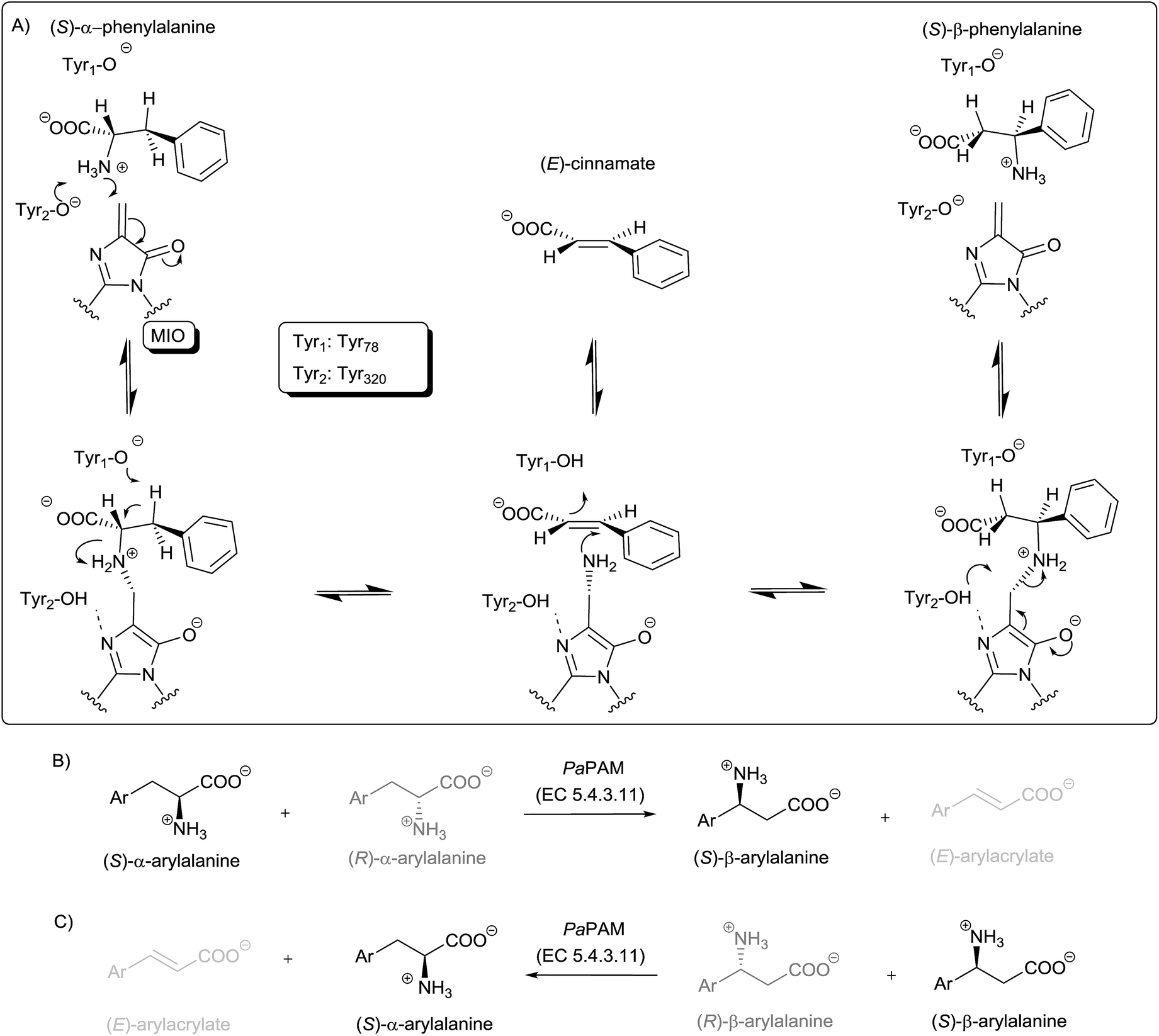 | ||
| Fig. 1 The proposed mechanism of action of PaPAM (A): starting from either direction with the cooperation of two catalytically essential tyrosine residues (Tyr1 and Tyr2) and the MIO prosthetic group, N-MIO intermediates of α- or β-phenylalanine are formed which interconvert by α ↔ β migration of the amino group involving a postulated (E)-cinnamate intermediate. PaPAM-catalysed kinetic resolutions starting from (B) racemic α-arylalanines or (C) racemic β-arylalanines result in the opposite enantiomers as products vs. unreacted enantiomers. | ||
The broad range of aromatic and heteroaromatic amino acids tolerated as substrates by these enzymes was also exploited for the preparation of a wide range of non-natural aryl and heteroaryl α- and β-amino acids.13
PAM from Taxus canadensis (TcPAM) forming (R)-β-phenylalanine was used for the partial biotransformation of (S)-α-phenylalanine and its derivatives into their (R)-β-isomer,5,14 while the closely related Taxus chinensis (TchPAM) as biocatalyst was employed in the enantioselective ammonia addition to (E)-cinnamate producing a mixture of enantiopure (S)-α- and (R)-β-phenylalanine.15 In a later study, significant shift of the regioisomeric preference towards the β-isomers was achieved by site directed mutagenesis.16 Wanninayake et al. have exploited some unnatural amino acids as amino group donors.17 The amino group of these substrates was transferred by TcPAM intermolecularly to another arylacrylate skeleton to form mixtures of α- and β-arylalanines.
Acting on α-phenylalanines the (S)-isomer-preferring PAM (EC 5.4.3.11) forms (S)-β-phenylalanine transposing exclusively the amino group of the (S)-α-phenylalanine to give (S)-β-phenylalanine (Fig. 1B).6 Among the known members of the PAM family producing (S)-β-phenylalanine are AdmH from Pantoea agglomerans (PaPAM)6 and EncP from Streptomyces maritimus (SmPAM).7 These enzymes have important applications in the preparation of the antibiotic andrimid,18 further various chiral phenylalanine derivatives19 as well as in the synthesis of the anticancer drug Taxol.20
The crystal structure of PaPAM complexed with phenylalanine to its active site, supported a reaction mechanism proceeding through two N-MIO containing intermediates.6 This was also in agreement with QM/MM calculations on TAL21 and PAL22 supporting ammonia elimination via an N-MIO intermediate and suggesting the formation of similar N-MIO complexes as a common feature of the mechanism for all MIO-enzymes.21 According to these propositions, the steps starting from either α- or β-phenylalanine are quite similar such as: (i) formation of a covalent enzyme–substrate complex via Michael addition of the amino group of the substrate onto MIO, (ii) ammonia elimination from the covalent N-MIO intermediate resulting in a cinnamate binding intermediate state (Fig. 1A). The mechanism proceeds further with (iii) ammonia re-addition and (iv) the release of the product. Occasionally, cinnamic acid can appear as a by-product (Fig. 1A), supposedly due to intermittent opening of the Tyr78-containig loop resulting in a leak from the main cycle at the cinnamate binding intermediate state.
SmPAM, described earlier as a lyase, has been shown lately to be closely related to PaPAM (63% overall sequence identity and 76% sequence similarity).7 More recently, Weise et al. investigated SmPAM in the context of ammonia addition to several aryl-substituted (E)-cinnamic acid analogues.23 They found that SmPAM converted a range of arylacrylates to a mixture of (S)-α- and (S)-β-arylalanines. The enzyme exhibited variable regioselectivity, much affected by ring substituents, but introduction of certain active site mutations could shift regioselectivity in either direction. However, in SmPAM-catalysed isomerization of (S)-α- and (S)-β-arylalanines the enantioselectivity was incomplete in many cases.23
Substrate specificity of PaPAM was tested with a wide range of aromatic and heteroaromatic (S)-α-arylalanines.19 Electronic and steric effects of substituents at the aromatic ring significantly influenced both catalytic efficiency and the formation of arylacrylates as by-products. It was observed that 3-substituted (S)-α-phenylalanines were transformed faster than the 2- or 4-substituted isomers. In order to explain these observations, computational analysis of substrate–PaPAM structural interactions was performed by substrate docking studies.19 Recently it was shown that recombinant whole cell E. coli expressing PaPAM could also produce enantiopure (S)-β-arylalanines from (S)-α-arylalanines.24 Worth noting, that PaPAM failed to catalyse the transformation of several 2-substituted (S)-α-phenylalanines studied.19,24
Results and discussion
The time course of the reaction (ESI‡ material) showed that after a relatively short time period (2–6 hours) an equilibrium state is reached in case of both enzymatic reactions, using rac-β-phenylalanines (Fig. 1C) or rac-α-phenylalanines (Fig. 1B) as substrates. Therefore the results after 20 h show the final product distribution near to the equilibrium state, after longer reaction times further product formation was not observed.Transformation of (±)-α-arylalanines (rac-1a–l) with PaPAM
According to previous results,19 PaPAM failed to catalyse transformation of 2-substituted (S)-α-phenylalanines bearing large electron withdrawing substituents. Our study starting from (±)-α-arylalanines rac-1a–l (Fig. 2A) confirmed the validity of this observation also for the racemic mixtures (Table 1).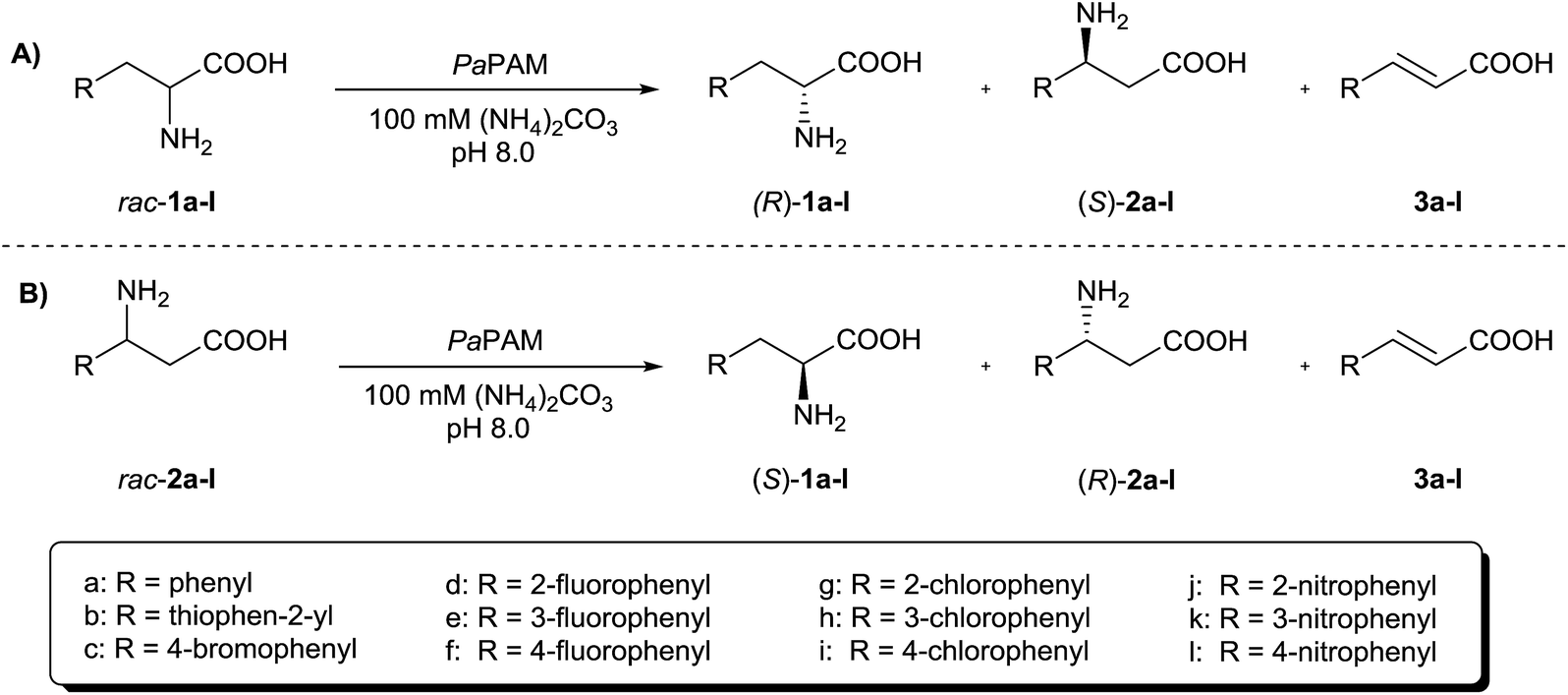 | ||
| Fig. 2 PaPAM-catalysed transformation of (A) (±)-α-arylalanines rac-1a–l and (B) (±)-β-arylalanines rac-2a–l. | ||
| Substrate (Ar) | x 1 | x (S)- 2 a | x 3 |
|---|---|---|---|
| a ee > 98% when not stated otherwise. b No reaction was observed. c Not observed. d x 1 , x(S)-2 and x3 represent the relative molar fractions of the reaction components as determined by 1H-, 19F-NMR measurements. | |||
| rac-1a (phenyl) | 0.66 | 0.28 | 0.04 |
| rac-1b (thiophen-2-yl) | 0.74 | 0.15 | 0.11 |
| rac-1c (4-bromophenyl) | 0.82 | 0.18 | c |
| rac-1d (2-fluorophenyl) | 0.8 | 0.07 | 0.13 |
| rac-1e (3-fluorophenyl) | 0.77 | 0.19 | 0.04 |
| rac-1f (4-fluorophenyl) | 0.77 | 0.20 | 0.03 |
| rac-1g (2-chlorophenyl) | 1.00b | b | b |
| rac-1h (3-chlorophenyl) | 0.79 | 0.2 | 0.02 |
| rac-1i (4-chlorophenyl) | 0.94 | 0.06 | c |
| rac-1j (2-nitrophenyl) | 1.00b | b | b |
| rac-1k (3-nitrophenyl) | 0.78 | 0.22 | c |
| rac-1l (4-nitrophenyl) | 0.94 | 0.06 | c |
In the presence of PaPAM, no product could be detected with (±)-α-(2-chlorophenyl)alanine (rac-1g) or (±)-α-(2-nitrophenyl)alanine (rac-1j), while with (±)-α-(2-fluorophenyl)alanine (rac-1d) being substituted with the smallest halogen atom moderate mutase-activity was observed resulting in the formation of enantiopure (S)-β-(2-fluorophenyl)alanine [(S)-2d].
Conversion of the (±)-3-substituted-α-phenylalanines (rac-1e, h, k) with PaPAM within 20 h was moderately faster than that of the 2-substituted substrate rac-1d, but still slower than with (±)-α-phenylalanine (rac-1a).
Comparison of the conversions of the (±)-4-substituted-α-phenylalanines (rac-1c, f, i, l) with PaPAM for 20 h indicated that with (±)-4-fluoro- (rac-1f) and (±)-4-bromo-α-phenylalanine (rac-1c) about 1.2 times higher conversions were attained than with (±)-4-chloro-(rac-1i) or (±)-4-nitro-α-phenylalanine (rac-1l).
Reactions of (±)-α-arylalanine bearing thiophen-2-yl group as a small heteroaromatic moiety (rac-1b) with PaPAM were also investigated. It was found that the conversion of (±)-α-(thiophen-2-yl)alanine (rac-1b) was similar to (±)-α-phenylalanine (rac-1a).
In an earlier study with (S)-1a, negligible ammonia-lyase activity was predicted for PaPAM at low temperatures (under 30 °C).7 In another study19 and also in our present one, however, formation of significant amount of (E)-arylacrylates was observed in many cases (3a, b, d, e, f, h, l in Table 1) indicating substantial lyase-activity of PaPAM. Note that with (±)-4-bromo- (rac-1c), (±)-4-chloro (rac-1i) and (±)-3-nitro-α-phenylalanine (rac-1k) as substrates no such activity was observed. Relatively high amounts of arylacrylates were formed in reactions with substrates bearing the smallest aromatic moieties i.e. with (±)-α-phenylalanine (rac-1a), (±)-α-(thiophen-2-yl)alanine (rac-1b) and (±)-2-fluoro-α-phenylalanine (rac-1d).
Results with the (±)-α-arylalanines (rac-1a–l) demonstrated that mutase and/or lyase activity of PaPAM was much affected by the nature of the aromatic moiety of the substrates. Importantly, high enantiopurity of the products [>98% ee for (S)-2a–f, h, i, k, l] indicated high enantioselectivity of the PaPAM-catalysed isomerization in the α → β-direction. The observed high stereoselectivity of the PaPAM-catalysed isomerization of the (S)-α- and (S)-β-arylalanines was a major advantage compared to the incomplete stereoselectivity of SmPAM-catalysed isomerizations.23
Transformation of (±)-β-arylalanines (rac-2a–l) with PaPAM
In order to explore the potential of kinetic resolutions of (±)-α- and β-arylalanines for the preparation of antipodal products, we extended our study to the reactions of (±)-β-arylalanines rac-2a–l (Fig. 2B and Table 2). The similar time course profiles of the product formation in PaPAM catalyzed reactions from (S)-β-phenylalanine and rac-β-phenylalanine (ESI‡ material) supported that the unreactive (R)-β-phenylalanine did not act as a significant inhibitor. In contrast to the (±)-2-substituted α-phenylalanines with large substituents (rac-1g, j), which were apparently not accepted as substrates by PaPAM, all the (±)-2-substituted β-phenylalanines in the present study (rac-1d, g, j) were smoothly transformed. On the other hand, sluggish or no conversion was observed with PaPAM using as substrates (±)-3- and (±)-4-substituted β-phenylalanines bearing bulky electron withdrawing substituents (rac-2c, h, i, k, l).| Substrate (Ar) | x 2 | x (S)- 1 a | x 3 |
|---|---|---|---|
| a ee > 98% when not stated otherwise. b No reaction was observed. c ee = 92%. d Not observed. e x 2 , x(S)-1 and x3 represent the relative molar fractions of the reaction components as determined by 1H-, 19F-NMR measurements. | |||
| rac-2a (phenyl) | 0.70 | 0.29 | 0.01 |
| rac-2b (thiophen-2-yl) | 0.73 | 0.22 | 0.04 |
| rac-2c (4-bromophenyl) | 0.91 | 0.08 | 0.01 |
| rac-2d (2-fluorophenyl) | 0.63 | 0.35 | 0.02 |
| rac-2e (3-fluorophenyl) | 0.67 | 0.25 | 0.08 |
| rac-2f (4-fluorophenyl) | 0.76 | 0.23 | 0.01 |
| rac-2g (2-chlorophenyl) | 0.54 | 0.40 | 0.06 |
| rac-2h (3-chlorophenyl) | 1.00b | b | b |
| rac-2i (4-chlorophenyl) | 1.00b | b | b |
| rac-2j (2-nitrophenyl) | 0.85 | 0.15c | d |
| rac-2k (3-nitrophenyl) | 1.00b | b | b |
| rac-2l (4-nitrophenyl) | 1.00b | b | b |
The conversions of (±)-β-(thiophen-2-yl)alanine (rac-2b) and (±)-3-fluoro-β-phenylalanine (rac-2e) with PaPAM were higher than that of (±)-β-phenylalanine (rac-2a). The (±)-4-fluoro-β-phenylalanine (rac-2f) was converted similarly as (±)-β-phenylalanine (rac-2a), while (±)-4-bromo-β-phenylalanine (rac-2c) was transformed to the α-isomer (S)-1c and a small amount of 4-bromocinnamate (3c) at significantly lower conversion than rac-2a. In all cases, except for (±)-2-nitro-β-phenylalanine (rac-2j), reactions catalysed by PaPAM proceeded with the formation of the enantiopure (S)-α-isomer [(S)-3a–g] and some arylacrylate (3a–g). Transformation of (±)-2-nitro-β-phenylalanine (rac-2j) was an exception owing to the absence of the by-product (3j) and incomplete stereoselectivity (ee(S)-1j = 92%).
Effects of pH and ammonia concentration on the PaPAM-catalysed isomerization of (±)-α- and β-(thiophen-2-yl)alanine (rac-1b and rac-2b)c
Prompted by the fact that conversions from (±)-α- and (±)-β-(thiophen-2-yl)alanine (rac-1b and rac-2b) with PaPAM were similar to those from the natural substrates i.e. (±)-α- and β-phenylalanine (rac-1a and rac-2a) but more of the by-product [(E)-3-(thiophen-2-yl)acrylate, 3b] was formed (Tables 1 and 2), transformations of these substrates were studied in more detail by varying pH or ammonia concentration. An alteration of pH of the buffer solution in the range of 7–9 [at 100 mM (NH4)2CO3] was indifferent to conversion (data not shown), unlike changing the (NH4)2CO3 concentration in the range of 50–1000 mM (at pH 8) which significantly influenced product compositions (Table 3).| Substrate | c (NH4)2CO3 (mM) | x 1b | x 2b | x 3b |
|---|---|---|---|---|
| a x 1b , x2b and x3b represent the relative molar fractions of the reaction components as determined by 1H-NMR measurements. | ||||
| rac-1b | 50 | 0.68 | 0.17 | 0.15 |
| rac-1b | 100 | 0.68 | 0.17 | 0.15 |
| rac-1b | 200 | 0.72 | 0.16 | 0.12 |
| rac-1b | 1000 | 0.76 | 0.12 | 0.12 |
| rac-2b | 50 | 0.19 | 0.76 | 0.05 |
| rac-2b | 100 | 0.27 | 0.69 | 0.04 |
| rac-2b | 200 | 0.23 | 0.75 | 0.02 |
| rac-2b | 1000 | 0.13 | 0.84 | 0.03 |
Analysis of the PaPAM-catalysed reactions of (±)-α-(thiophen-2-yl)alanine (rac-1b) at various (NH4)2CO3 concentrations with 20 h incubation showed that increasing (NH4)2CO3 concentration above 100 mM resulted in decreasing conversion to both (S)-β-(thiophen-2-yl)alanine (S)-2b and the elimination product 3b. At 1000 mM (NH4)2CO3 concentration both (S)-β-(thiophen-2-yl)alanine (S)-2b and arylacrylate 3b formation dropped to 12%. Interestingly, when starting from (±)-β-(thiophen-2-yl)alanine (rac-2b), the best conversion to (S)-α-(thiophen-2-yl)alanine (S)-1b (27%) was achieved at 100 mM buffer concentration. At higher (NH4)2CO3 concentrations smaller conversions were observed.
Study of the dependence of the PaPAM-catalysed reactions of (±)-α- and β-(thiophen-2-yl)alanine (rac-1b and rac-2b, respectively) on (NH4)2CO3 concentration indicated that elevated ammonia concentrations lowered both the rate of isomerization and ammonia elimination as side reaction in both directions of the reaction.
Computational modelling of PaPAM-catalysed isomerizations
To rationalize the effect of substituents of aromatic and heteroaromatic (±)-α- and β-arylalanines (rac-1a–l and rac-2a–l) on PaPAM-catalysed isomerizations computational and statistical analysis was performed based on modelling and comparison of the N-MIO states (S)-1a–i, k, lN-MIO and (S)-2a–i, k, lN-MIO formed from the corresponding (S)-α- and (S)-β-arylalanines. Because of the experimentally observed stereospecificity of isomerizations, in the computations only the (S)-enantiomers were considered. Due to the mixed mechanism indicated by the incomplete enantioselectivity in the PaPAM-catalysed reaction of rac-2j, data calculated for the reactions of (S)-α- and (S)-β-2-nitrophenylalanine [(S)-1j and (S)-2j] were omitted from the comparison of reactivities.In molecular mechanics the bonded terms measure the strain compared to a hypothetical zero-point energy. Thus potential energies derived from molecular mechanics calculations cannot be compared directly if the molecular structures to be related do not have exactly the same atom connectivity. In this modelling study it was assumed that for all investigated compounds the common alanine part of α- and β-arylalanines underwent in the corresponding N-MIO intermediates similar structural changes6,21 (Fig. 1). Therefore, it was possible to compare the difference of the energies calculated for the N-MIO intermediates from the (S)-α- and (S)-β-arylalanines [(S)-1b–i, k, lN-MIO and (S)-2b–i, k, lN-MIO] relative to the corresponding values obtained for (S)-α- and (S)-β-phenylalanine [(S)-1aN-MIO and (S)-2aN-MIO], explicitly: (E(S)-2 − E(S)-1) − (E(S)-2a − E(S)-1a), thus cancelling the hypothetical terms representing the difference in heat of formation for the α- and β-alanine part (Table 4).
| Substrates (Ar) | Difference of conversionsa | Categoryb | Energy differencec (kcal mol−1) |
|---|---|---|---|
| a Difference of conversions is defined as: (1 − x2) − (1 − x1), where x1, x2 are the corresponding molar fraction values listed in Tables 1 and 2. b Categories based on regioisomeric preference are defined as follows: A: (1 − x2) − (1 − x1) < −0.09; E: −0.09 ≤ (1 − x2) − (1 − x1) ≤ 0.09; B: (1 − x2) − (1 − x1) > 0.09. c Energy difference of N-MIO states (S)-1b–lN-MIO and (S)-2b–lN-MIO is defined and normalized for (S)-1aN-MIO and (S)-2aN-MIO as: (E(S)-2 − E(S)-1) − (E(S)-2a − E(S)-1a). For compounds with different ring orientations in their lowest energy conformations of the α- and β-N-MIO intermediate states, energy values are selected for structures consistent with the reaction of lower or no conversion. Energy differences pertinent to the other direction are presented in parentheses. d Due to the mixed mechanism indicated by the incomplete stereoselectivity in the reaction of rac-2j with PaPAM, data for (S)-1jN-MIO and (S)-2jN-MIO were not included. | |||
| (S)-1, 2a (phenyl) | −0.05 | E | 0.0 |
| (S)-1, 2b (thiophen-2-yl) | −0.01 | E | 6.1 |
| (S)-1, 2c (4-bromophenyl) | −0.12 | A | −3.6 |
| (S)-1, 2d (2-fluorophenyl) | 0.19 | B | 1.8 (1.0) |
| (S)-1, 2e (3-fluorophenyl) | 0.09 | E | −4.1 |
| (S)-1, 2f (4-fluorophenyl) | 0.01 | E | 0.2 |
| (S)-1, 2g (2-chlorophenyl) | 0.46 | B | 3.1 (0.6) |
| (S)-1, 2h (3-chlorophenyl) | −0.16 | A | −3.0 |
| (S)-1, 2i (4-chlorophenyl) | −0.10 | A | −2.8 |
| (S)-1, 2j (2-nitrophenyl) | d | d | d |
| (S)-1, 2k (3-nitrophenyl) | −0.26 | A | −7.2 (−2.0) |
| (S)-1, 2l (4-nitrophenyl) | −0.11 | A | −6.7 |
Our molecular modelling studies based on an exhaustive search for possible conformations of the covalently bound N-MIO intermediates of the (S)-α- and (S)-β-arylalanines [(S)-1b–i, k, lN-MIO and (S)-2b–i, k, lN-MIO], indicated that unlike the reactions of the majority of the regioisomers [(S)-1, 2a–c, e, f, h–j, l; see Fig. 3A25 for (S)-1hN-MIO and (S)-2hN-MIO], in certain pairs of isomers [(S)-1, 2d, (S)-1, 2g and (S)-1, 2k] the arrangements of the aromatic ring in the lowest energy conformations of the enzyme-bound substrate complexes were dissimilar and differing by a 180° flip [see Fig. 3B for (S)-1gN-MIO and (S)-2gN-MIO]. Assuming that in the N-MIO complexes in the tightly packed active site of PaPAM there is no room for a flip neither of the complete arylacrylate molecule nor of the aromatic ring only, it can be predicted that in such cases one of the two N-MIO intermediate states is not in its lowest energy state. Thus the calculated lowest energy differences in such cases should be higher, corresponding to the allowed arrangements without involving a sterically hindered ring flip (Table 4).
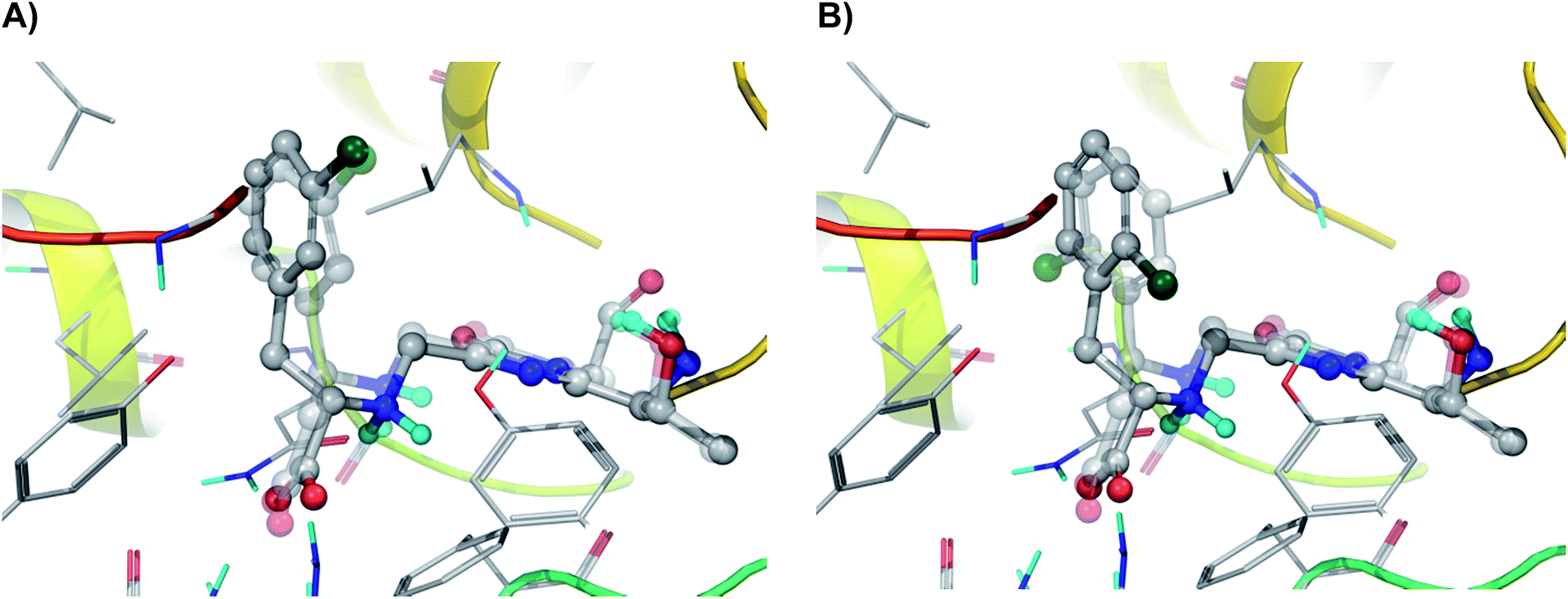 | ||
| Fig. 3 Comparison of the arrangements of regioisomeric N-MIO intermediates in their PaPAM-catalysed transformations. (A) N-MIO intermediates of (S)-α-(3-chlorophenyl)alanine (solid model) and of (S)-β-(3-chlorophenyl)alanine (transparent model) [(S)-1hN-MIO and (S)-2hN-MIO]; (B) the N-MIO intermediates of (S)-α-(2-chlorophenyl)alanine (solid model) and (S)-β-(2-chlorophenyl)alanine (transparent model) [(S)-1gN-MIO and (S)-2gN-MIO]. Panel (A) depicts a case where the lowest energy conformations of the two regioisomers adopt similar orientations of the aromatic ring. Panel (B) illustrates a case where in the lowest energy conformations of the two regioisomers the aromatic rings occupy different orientations (differing by a flip of 180°). Images were created using PyMOL.25 | ||
Statistical analysis of the experimental and computational data revealed that compounds (S)-1, 2a–i, k, l could be classified into three categories (A, E and B, in Table 4) based on the difference of conversions [defined as: (1 − x2) − (1 − x1), where x1, x2 are the corresponding molar fraction values listed in Tables 1 and 2]. The three categories: A: (1 − x2) − (1 − x1) < −0.09; E: −0.09 ≤ (1 − x2) − (1 − x1) ≤ 0.09 and B: (1 − x2) − (1 − x1) > 0.09 could be correlated with the regioisomeric preferences (Table 4, Fig. 4). One-way ANOVA test – treating the categories (Table 4) as the independent and the conversion differences (Tables 1 and 2) as the dependent variables – indicated significant difference between categories A and B at the α = 0.050 level. In addition, further nonparametric tests – having lower statistical power than parametric methods but without the requirement of normal distribution of data – were also performed. The Kruskal–Wallis ANOVA and Mann–Whitney U tests resulted only somewhat higher p values (0.100 and 0.053, respectively) than the threshold. Moreover, the median test showed also significant differences between the categories. Overall, the statistical tests supported the finding that categories A and B are truly separate.
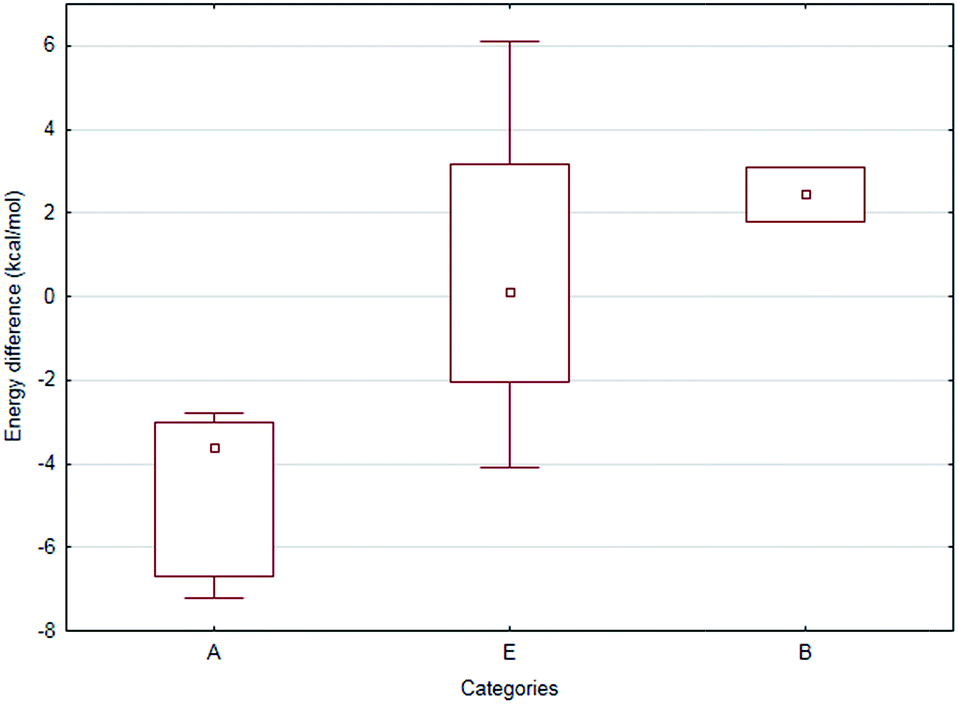 | ||
| Fig. 4 Box–whisker plot depicting the energy differences of the corresponding N-MIO intermediates [(S)-1a–i, k, lN-MIO and (S)-2a–i, k, lN-MIO] as a function of the catalytic activity categories (A, B and E). Legend – square: median; boxes: span between the lower and upper quartile, whiskers: span of the entire set. | ||
The computational study extended with statistical analysis revealed that the cases when one of the regioisomers was converted much faster (or is the only one to be converted) the energetics of the regioisomeric N-MIO-type enzyme–substrate complexes was one of the most important factors governing the outcome of the reaction. In the case of the intermediate group (category E) involving small aromatic moieties contribution of the steric effects are much less pronounced.
The computational results listed in Table 4 suggest that the reactions of 2-substituted α-phenylalanines [(S)-1d, g and (S)-2d, g: category B in Table 4 and Fig. 4] are strongly disfavoured because the energy calculated for the α-N-MIO intermediates [(S)-1d, gN-MIO] is much lower than that calculated for the corresponding β-N-MIO intermediates [(S)-2d, gN-MIO]. This was in full agreement with experimental observations indicating slow or no reaction from the (±)-2-substituted α-phenylalanines (rac-1d, g, j) with PaPAM, in contrast to high conversions from 2-substituted (±)-β-phenylalanines (rac-2d, g, j) under the same conditions. The situation for the 3- and 4-substituted phenylalanines with bulky substituents [(S)-1c, h, i, k, l and (S)-2c, h, i, k, l: category A in Table 4 and Fig. 4] was the opposite. In good correlation with the experimental regioisomeric preferences, much lower energies were calculated for the β-N-MIO intermediates [(S)-2c, h, i, k, lN-MIO] than for the corresponding α-N-MIO intermediates [(S)-1c, h, i, k, lN-MIO]. This could explain sluggish or no conversion of β-arylalanines containing bulky substituents at positions 3 or 4 (rac-2c, h, i, k, l) with PaPAM and much higher conversions of the α compounds (rac-1c, h, i, k, l) under the same conditions. In addition, the energy differences corresponding to the direction of reaction with higher conversions and assuming a disallowed flip of the aromatic ring (Table 4, values in parentheses) showed remarkable differences compared to the sterically non-restricted values and the hindered flip of the aromatic ring further amplified the difference in reactivity mentioned before. Because of the hindered flip, in case of certain regioisomeric amino acid pairs the aromatic ring in a given N-MIO intermediate adopts different conformation depending on whether the actual N-MIO intermediate is at the substrate side [step (ii) in our postulated mechanism, Fig. 1A] or at the product side [step (iii) in our postulated mechanism, Fig. 1A].
Conclusion
According to our results, regioselectivity and activity of PaPAM towards α- and β-arylalanines are mainly influenced by the nature of the aromatic moiety of the substrates. PaPAM catalyses the synthesis of the corresponding (S)-β-enantiomer from racemic 3- and 4-substituted α-phenylalanines more smoothly than that starting from racemic 2-substituted α-phenylalanines, the latter being poor or no substrates of the enzyme. Contrarily, racemic 2-substituted β-phenylalanines were good substrates and provided the (S)-α-enantiomers smoothly while the racemic 3- or 4-substituted β-phenylalanines were almost no substrates. Importantly, in all but one case (for racemic 2-nitro-β-phenylalanine), the isomerizations were stereospecific giving mixture of unreacted (R)-α- or (R)-β-arylalanines with an enantiomeric excess depending on conversion, and enantiopure (S)-α- or β-arylalanines as products, along with various amounts of arylacrylate as by-product. Computational and statistical analysis revealed significant correlation between the energetics of the N-MIO intermediate states forming from the (S)-α- or β-arylalanines and the regioisomeric preferences of PaPAM in case of substrates with bulky electron withdrawing substituents on the aromatic ring. In several cases “hysteresis” was postulated: the conformation (thus energy) of a given regioisomeric N-MIO intermediate formed from the given regioisomeric substrate differed from that conformation which resulted in the given regioisomer as product.Experimental
Reagents and analytical methods
The starting materials were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Carl Roth (Karlsruhe, Germany) and used without purification. Solvents were purified and dried by standard methods. The racemic amino acids rac-1a–lrac-2a–l and the α,β-unsaturated acids 3a–l were synthesized using published methods.26The 1H- and 19F-NMR spectra were recorded at 21 °C on Bruker spectrometers operating at 400 MHz, 101 MHz and 600 MHz, 151 MHz, respectively. Enantiomer separations were obtained on Agilent 1260 HPLC instrument using either Chiralpak® ZWIX(+) (4 mm × 250 mm) column and a mixture of methanol (containing 100 mmol L−1 formic acid and 50 mmol L−1 diethylamine), acetonitrile and water in proportion of 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49:2 (v/v/v) as eluent at a flow rate of 1 mL min−1 or Crownpak® CR-I(+) column (150 × 3.0 mm × 5 μm) and a mixture of HClO4 solution (3.6 g L−1, pH 1.5): acetonitrile as mobile phase at a flow rate of 0.4 mL min−1. NMR spectra and HPLC data are presented as ESI‡ material.
:49:2 (v/v/v) as eluent at a flow rate of 1 mL min−1 or Crownpak® CR-I(+) column (150 × 3.0 mm × 5 μm) and a mixture of HClO4 solution (3.6 g L−1, pH 1.5): acetonitrile as mobile phase at a flow rate of 0.4 mL min−1. NMR spectra and HPLC data are presented as ESI‡ material.
Expression and purification of PaPAM
The gene of PAM from Pantoea agglomerans (encoding 541 AAs – Uniprot code: Q84FL5) was optimized to the codon usage of E. coli. The 1626 bps long synthetic gene was produced and cloned into pET-19b vector using the XhoI and Bpu1102I cloning sites. The recombinant PaPAM carrying an N-terminal (His)10-tag was produced in E. coli BL21(DE3)pLysS cells. For the expression step a colony of the transformed plasmid was grown overnight at 37 °C in 5 mL of LB medium containing carbenicillin (50 μg mL−1) and chloramphenicol (30 μg mL−1). The overnight culture was added to LB medium (0.5 L) in an Erlenmeyer flask and grown at 37 °C until OD600 reached 0.7–0.8. Then the temperature was lowered to 25 °C and the cells were induced by the addition of IPTG (1 mM). The culture was shaken at 220 rpm at 25 °C for 19 h longer. All of the subsequent procedures were carried out in an ice-bath. The cells were harvested by centrifugation (25 min, 5000 × g) and re-suspended in 50 mL of lysis buffer (150 mM NaCl, 50 mM TRIS, pH 7.5) supplemented with DNAse, RNAse, lysozyme, PMSF (2 mM) and an EDTA-free protease-inhibitor cocktail. The cells were then lysed by sonication and the cell debris was removed by centrifugation (10000 × g, 30 min).
The proteins were purified on a column filled with nickel–nitrilotriacetic acid agarose gel (Ni–NTA) following the manufacturer's protocol.27 The expressed protein was eluted from the column with imidazole (500 mM in low salt buffer, pH 7.5). The purity of the protein in the eluted fractions was verified by SDS-PAGE analysis. The protein fractions were dialyzed against phosphate-buffered saline (50 mM) at 4 °C followed by concentration by centrifugal ultrafiltration (using vertically-oriented ultrafiltration membrane VIVASPIN 10000 MWCO, 5000 × g, 4 °C, to final concentration of 3–5 mg mL−1). The concentration of PaPAM in the final solutions was determined by Bradford's method.28
PaPAM-catalysed biotransformations of (±)-α- and β-arylalanines
Into the solution of the substrate (rac-1a–f, h, i, k, l and rac-2a–g, j, 4 mg) in (NH4)2CO3 buffer (100 mM, pH 8.0, 2 mL), PaPAM (1.6 mg) was added and the reaction mixture was stirred at room temperature for 20 h. For HPLC measurements reaction samples (30 μL) were taken and the enzyme was precipitated with 30 μL MeOH. After filtration, the samples were diluted with the corresponding mobile phase and injected on HPLC.Prior to 1H- and 19F-NMR investigations the reaction was quenched with methanol, followed by filtration and evaporation of the solvent in vacuum. Deuterated sodium hydroxide (2% NaOD) solution was added and the spectrum (ESI‡) was recorded at room temperature.
Biotransformation of (±)-α- or β-(thiophen-2-yl)alanine (rac-1b or rac-2b) under various conditions
Molecular modelling of the covalent enzyme–substrate N-MIO complexes in PaPAM
The homotetrameric X-ray structure of PaPAM [PDB ID: 3UNV]6 was completed and adjusted using the Protein Preparation Wizard29 in four steps: (i) hydrogen atoms were added and bond orders were assigned, (ii) artefacts of the protein crystallization procedure were removed, except for two phosphate ions, (iii) hydrogen bond network, tautomeric states, side chain conformations of selected amino acids and ionization states were determined and optimized corresponding to the experimental assay conditions and (iv) a constrained minimization was performed. Protein pKa were predicted using PROPKA.30 In the further modelling process for the N-MIO intermediates, in accordance with to the proposed mechanism (Fig. 1), Tyr78 was set deprotonated and Tyr320 protonated. Further details on the computational methods and the model will be published in a forthcoming paper.The refined and completed X-ray structure served as a starting point to create an overall protein model corresponding to the experimental assay conditions. The buffer solution solvated model was created by the Desmond program suite.31 The PaPAM model was solvated explicitly with water and additional ions were added with respect to the experimental assay conditions. The buffer solvated model was then equilibrated with a slightly modified default equilibration protocol, applying harmonic constraints to the Cartesian coordinates of protein heavy atoms. A spherical model of the active site with a radius of 27 Å and centred on the exocyclic methylene carbon of the MIO prosthetic group of chain C was cut off and capped with acetyl and N-methylamino groups.
N-MIO type covalent complexes of our substrates (S)-1a–l and (S)-2a–lwere created by our induced-fit covalent docking protocol. This involved the creation of initial conformations of compounds (S)-1a–l and (S)-2a–l by docking with Glide program suite32 into a modified and artificially enlarged active site in which the residues Leu216, Ile219, Leu104, Val108, Met84, Leu421, Leu171, and Phe428 were exchanged to Ala residues, further the MIO prosthetic group was reduced to Thr + Gly and three water molecules in the active site were removed.
After having docked into the enlarged active site, all side chains and the MIO group were restored, a covalent bond between the nitrogen atom of the amino group and the exocyclic carbon of MIO was created, the covalently bound ligands and the residues in close proximity to them were minimized and finally, redundant conformations were eliminated with MacroModel.33 During restoring the mutated side-chains, conformations of several active site residues were predicted with Prime.34
After replacing the three active site water molecules removed earlier, a final minimization in the 6 Å proximity of the covalently bound ligand using Prime34 resulted in the final models and energies. OPLS2005 force field was applied in all molecular mechanics calculations and simulations.
Statistical tests (one-way ANOVA, Kruskal–Wallis ANOVA, median test and Mann–Whitney U tests) were carried out and Fig. 4 was created using Statistica.35 Non-significant Levene and Shapiro–Wilk tests justified the use of one-way ANOVA. The probability value of type I error (α) was chosen to be 0.05 in all the cases.
Acknowledgements
AV and GB contributed equally to this work. AV and NB thank the financial support from the Sectoral Operational Program for Human Resources Development 2007–2013, co-financed by the European Social Fund (projects POSDRU/159/1.5/S/137750 and POSDRU/159/1.5/S/132400). The financial support provided by the Collegium Talentum Research Program to NB and LP is also acknowledged. CP thanks for financial support from the Romanian National Authority for Scientific Research, CNCS – UEFISCDI (PN-II-IDPCE-2011-3-0799). LP thanks for financial support from Hungarian OTKA Foundation (NN-103242), from the Hungarian Research and Technology Innovation Fund (KMR 12-1-2012-0140) and from the New Hungary Development Plan (TÁMOP-4.2.1/B-09/1/KMR-2010-0002). Licensing of the Schrödinger Suite software package was financed by the Hungarian OTKA Foundation (K 108793). LP and CP thank the support from COST Action CM1303 (SysBiocat). We thank Prof. Mihály Nógrádi (BME, Budapest) and Dr Károly Héberger (RCNS HAS, Budapest) for helpful discussions.Notes and references
- (a) F. Fülöp, T. A. Martinek and G. K. Tóth, Chem. Soc. Rev., 2006, 35, 323–334 RSC; (b) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed.
- L. Kiss and F. Fülöp, Chem. Rev., 2014, 114, 1116–1169 CrossRef CAS PubMed.
- F. van Rantwijk and R. A. Sheldon, Tetrahedron, 2004, 60, 501–519 CrossRef CAS.
- J. Altenbuchner, M. Siemann-Herzberg and C. Syldatk, Curr. Opin. Biotechnol., 2001, 12, 559–563 CrossRef CAS PubMed.
- L. Feng, U. Wanninayake, S. Strom, J. Geiger and K. D. Walker, Biochemistry, 2011, 50, 2919–2930 CrossRef CAS PubMed.
- S. Strom, U. Wanninayake, N. D. Ratnayake, K. D. Walker and J. H. Geiger, Angew. Chem., Int. Ed., 2012, 51, 2898–2902 CrossRef CAS PubMed.
- C. Chesters, M. Wilding, M. Goodall and J. Micklefield, Angew. Chem., Int. Ed., 2012, 51, 4344–4348 CrossRef CAS PubMed.
- (a) C. V. Christianson, T. J. Montavon, S. G. Van Lanen, B. Shen and S. D. Bruner, Biochemistry, 2007, 46, 7205–7214 CrossRef CAS PubMed; (b) D. Krug and R. Müller, ChemBioChem, 2009, 10, 741–750 CrossRef CAS PubMed.
- M. J. MacDonald and G. B. D'Cunha, Biochem. Cell Biol., 2007, 85, 273–282 CrossRef CAS PubMed.
- J. A. Kyndt, T. E. Meyer, M. A. Cusanovich and J. J. Van Beeumen, FEBS Lett., 2002, 512, 240–244 CrossRef CAS PubMed.
- (a) T. F. Schwede, J. Rétey and G. E. Schulz, Biochemistry, 1999, 38, 5355–5361 CrossRef CAS PubMed; (b) M. Baedecker and G. E. Schulz, Eur. J. Biochem., 2012, 269, 1790–1797 CrossRef.
- (a) H. A. Cooke and S. D. Bruner, Biopolymers, 2010, 93, 802–810 CrossRef CAS PubMed; (b) V. Kapatral, I. Anderson, N. Ivanova, G. Reznik, T. Los, A. Lykidis, A. Bhattacharyya, A. Bartman, W. Gartner, G. Grechkin, L. Zhu, O. Vasieva, L. Chu, Y. Kogan, O. Chaga, E. Goltsman, A. Bernal, N. Larsen and R. Overbeek, J. Bacteriol., 2002, 184, 2005–2018 CrossRef CAS PubMed; (c) P. C. Wu, T. A. Kroening, P. J. White and K. E. Kendrick, J. Bacteriol., 1992, 174, 1647–1655 CrossRef CAS PubMed.
- (a) N. J. Turner, Curr. Opin. Chem. Biol., 2011, 15(2), 234–240 CrossRef CAS PubMed; (b) L. Poppe, C. Paizs, K. Kovács, F. D. Irimie and B. Vértessy, Methods Mol. Biol., 2012, 794, 3–19 CrossRef CAS PubMed; (c) M. M. Heberling, B. Wu, S. Bartsch and D. B. Janssen, Curr. Opin. Chem. Biol., 2013, 17(2), 250–260 CrossRef CAS PubMed.
- K. Klettke, S. Sanyal, W. Mutatu and K. D. Walker, J. Am. Chem. Soc., 2007, 129, 6988–6989 CrossRef CAS PubMed.
- B. Wu, W. Szymanski, P. Wietzes, S. Wildeman, G. J. Poelarends, B. L. Feringa and D. B. Janssen, ChemBioChem, 2009, 10, 338–344 CrossRef CAS PubMed.
- B. Wu, W. Szymanski, G. G. Wybenga, M. M. Heberling, S. Bartsch, S. Wildeman, G. J. Poelarends, B. L. Feringa, B. W. Dijkstra and D. B. Janssen, Angew. Chem., Int. Ed., 2012, 51, 482–486 CrossRef CAS PubMed.
- U. Wanninayake, Y. DePorre, M. Ondari and K. D. Walker, Biochemistry, 2011, 50, 10082–10090 CrossRef CAS PubMed.
- N. D. Ratnayake, U. Wanninayake, J. H. Geiger and K. D. Walker, J. Am. Chem. Soc., 2011, 133, 8531–8533 CrossRef CAS PubMed.
- N. D. Ratnayake, N. Liu, L. A. Kuhn and K. D. Walker, ACS Catal., 2014, 4, 3077–3090 CrossRef CAS.
- K. D. Walker, K. Klettke, T. Akiyama and R. Croteau, J. Biol. Chem., 2004, 279, 53947–53954 CrossRef CAS PubMed.
- S. Pilbák, Ö. Farkas and L. Poppe, Chem.–Eur. J., 2012, 18, 7793–7802 CrossRef PubMed.
- D. Weiser, L. C. Bencze, G. Bánóczi, F. Ender, R. Kiss, E. Kókai, A. Szilágyi, B. G. Vértessy, Ö. Farkas, C. Paizs and L. Poppe, ChemBioChem, 2015, 16, 2283–2288 CrossRef CAS PubMed.
- N. J. Weise, F. Parmeggiani, S. T. Ahmed and N. J. Turner, J. Am. Chem. Soc., 2015, 137, 12977–12983 CrossRef CAS PubMed.
- N. D. Ratnayake, C. Theisen, T. Walker and K. D. Walker, J. Biotechnol., 2016, 217, 12–21 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC, New York, NY, USA Search PubMed.
- (a) C. Paizs, A. Katona and J. Rétey, Chem.–Eur. J., 2006, 12, 2739–2744 CrossRef CAS PubMed; (b) C. Y. K. Tan and D. F. Weaver, Tetrahedron, 2002, 58, 7449–7461 CrossRef CAS.
- Quiagen, The QIAexpressionist – A handbook for high-level expression and purification of 6xHis-tagged proteins, Quiagen, Valencia, CA, USA, 5th edn, 2003 Search PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- (a) Protein Preparation Wizard 2015-2: Epik, Version 2.4, Impact, Version 5.9 Search PubMed; (b) G. M. Satry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- M. H. M. Olsson, C. R. Søndergard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- (a) Desmond Molecular Dynamics System, Version 4.2, D. E. Shaw Research, New York, NY, USA Search PubMed; (b) Maestro-Desmond Interoperability Tools, Version 4.2, Schrödinger, New York, NY, USA Search PubMed; (c) K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Proceedings of the ACM/IEEE Conference on Super-computing (SC06), Tampa, FL, 2006, November 11–17 Search PubMed.
- (a) Glide 2015, Version 6.7, Schrödinger, LLC, New York, NY, USA Search PubMed; (b) R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, D. E. Shaw, M. Shelley, J. K. Perry, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed; (c) R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed; (d) T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- MacroModel, version 10.8, Schrödinger, LLC, New York, NY, 2015 Search PubMed.
- Prime, Version 4.0, Schrödinger, LLC, New York, NY, USA Search PubMed.
- StatSoft, Inc. (2014). STATISTICA (data analysis software system), version 12, https://www.statsoft.com Search PubMed.
Footnotes |
| † This paper is dedicated to Professor Albert Eschenmoser on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02964g |
| This journal is © The Royal Society of Chemistry 2016 |