
Integrating error-prone PCR and DNA shuffling as an effective molecular evolution strategy for the production of α-ketoglutaric acid by l-amino acid deaminase†
Abstract
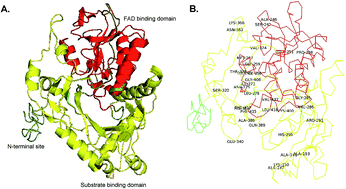
L-Amino acid deaminases (LAADs; EC 1.4.3.2) belong to a family of amino acid dehydrogenases that catalyze the formation of α-keto acids from L-amino acids. In a previous study, a whole cell biocatalyst with the L-amino acid deaminase (pm1) from Proteus mirabilis was developed for the one-step production of α-ketoglutarate (α-KG) from L-glutamic acid, and the α-KG titer reached 12.79 g L−1 in a 3 L batch bioreactor. However, the product α-KG strongly inhibited pm1 activity, and the titer of α-KG was comparatively lower than expected. Therefore, in this study, multiple rounds of error-prone polymerase chain reaction (PCR) and gene shuffling were integrated for the molecular engineering of pm1 to further improve the catalytic performance and α-KG titer. A variant (pm1338g4), which contained mutations in 34 amino acid residues, was found to have enhanced catalytic efficiency. In a batch system, the α-KG titer reached 53.74 g L−1 when 100 g of monosodium glutamate was used as a substrate. Additionally, in a fed-batch biotransformation system, the maximum α-KG titer reached 89.11 g L−1 when monosodium glutamate was continuously fed at a constant rate of 6 g L−1 h−1 (from 4 to 23 h) with an initial concentration of 50 g L−1. Analysis of the kinetics of the mutant variant showed that these improvements were achieved due to enhancement of the reaction velocity (from 56.7 μM min−1 to 241.8 μM min−1) and substrate affinity (the Km for glutamate decreased from 23.58 to 6.56 mM). A possible mechanism for the enhanced substrate affinity was also evaluated by structural modeling of the mutant. Our findings showed that the integration of error-prone PCR and gene shuffling was an effective method for improvement of the catalytic performance of industrial enzymes.
 Please wait while we load your content...
Please wait while we load your content...

