
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium nanoparticles generated in situ used as catalysts in carbonylative cross-coupling in aqueous medium†
P.
Wójcik
a,
M.
Mart
a,
S.
Ulukanli
b and
A. M.
Trzeciak
*a
aUniversity of Wrocław, Faculty of Chemistry, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: anna.trzeciak@chem.uni.wroc.pl
bDepartment of Chemistry, Faculty of Science and Letters, Osmaniye Korkut Ata University, 80000 Osmaniye, Turkey
First published on 4th April 2016
Abstract
Pd NPs, obtained in situ from imidazole complexes Pd(im)2Cl2 (im = methylimidazole, butylimidazole), efficiently catalyze the carbonylative Suzuki coupling in water at 80 °C and 1 atm of CO. In the catalytic system, used without any additional ligands, differently substituted diaryl ketones were obtained with yields of up to 100% in 1 h. Under slightly modified conditions, Pd NPs catalyze the alkoxycarbonylation of iodobenzenes to esters with high yield.
Introduction
Diaryl ketones are important building blocks present in a large number of natural products (cotton, papaveraldine) and anti-inflammatory drugs (suprofen, ketoprofen). They are also used in UV screens (sulisobenzone, oxybenzone), dyes and agrochemicals. The importance of diaryl ketones as intermediates in the manufacture of valuable industrial products stimulates searching more efficient and simple methods of their preparation.1–3Diaryl ketones can be prepared in the Friedel–Crafts acylation of substituted aromatic rings.4–6 This reaction requires excessive amounts of Lewis acid, harsh reaction conditions, and, besides, its regioselectivity is often low. The alternative is the acylation of aryl metal species with functional carboxylic acid derivatives.7–10 Here, organometallic compounds of tin, zinc, cadmium, magnesium, and lithium were used; however, the yield of the ketone product was also relatively low.11,12
A direct and more promising method leading to diaryl ketones is the carbonylative Suzuki coupling, the three-component coupling of aryl halide, boronic acid, and carbon monoxide, forming the final product in a single step. The majority of these reactions has been carried out in organic solvents with application of palladium complexes as catalysts.13–23 Carbonylative coupling of iodobenzene with tetraphenylborate was also described.24,25
Recently, the first examples of Pd NPs application to the carbonylative Suzuki coupling in toluene26 and in PEG-400 (ref. 27) have been reported. Nevertheless, to the best of our knowledge, the application of Pd NPs as catalysts for the carbonylative Suzuki coupling in water has not been reported yet.28
Therefore, our studies aimed at the development of the catalytic system for the carbonylative Suzuki coupling applying Pd NPs generated in situ in an aqueous medium. To this aim, a proper selection of the palladium precursor was the most important, and we selected Pd complexes with non-toxic imidazole ligands which show an excellent activity in the Suzuki coupling.29 An additional advantage is that Pd–imidazole complexes can be prepared with high yields according to a simple procedure; they are also easy to handle and stable in the presence of water and air.
Experimental
(1) Palladium complexes, Pd(1-BI)2Cl2 and Pd(1-MI)2Cl2, (1-BI = 1-butylimidazole, 1-MI = 1-methylimidazole) were obtained according to the literature method.29(2) Carbonylative Suzuki coupling reaction
(a) A 50 mL Schlenk flask was charged with aryl iodide (1 mmol), aryl boronic acid (1.2 mmol), Pd(1-BI)2Cl2 or Pd(1-MI)2Cl2 (0.01 mmol), and Na2CO3 (3 mmol). Then, 5 mL of distilled water was added. The Schlenk flask was evacuated and filled with CO. Under the balloon pressure of CO, the reaction mixture was stirred at 60 or 80 °C for 1–4 h. After that time, the Schlenk flask was cooled down, and the organic products were extracted with 3 × 7 mL of n-hexane (3 × 15 min with stirring). The extract was GC analyzed with mesitylene as the internal standard (0.076 mL, 5.46 × 10−4 mol).
(b) The palladium precursor (0.01 mmol), dihalobenzene (1 mmol), phenylboronic acid (2.2 mmol) and Na2CO3 (3 mmol) were placed in an autoclave under nitrogen atmosphere and 5 mL of distilled water was added. The autoclave was closed, purged three times with CO and then pressurized to 5 or 10 bar of CO. The reaction mixture was stirred at 80 °C for 1–18 h. After that time, the autoclave was cooled down, and the organic products were extracted with 3 × 7 mL of n-hexane (3 × 15 min with stirring). The extract was GC analyzed with mesitylene as the internal standard (0.076 mL, 5.46 × 10−4 mol).
Next, the solvents were evaporated, the crude product was purified by column chromatography on silica gel using hexane/ethyl acetate (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent, the corresponding diaryl ketone was obtained.
:1) as the eluent, the corresponding diaryl ketone was obtained.
(c) Three-phase test
The three-phase test was conducted in accordance with the literature procedure.30 In order to immobilize the aryl halide, 2-iodo-benzyl chloride (1.5 mmol) was dissolved in 20 mL of dry THF in a 50 mL Schlenk flask. 3-Aminopropyl-functionalized silica gel (1.5 g of silica gel functionalized with 1.5 mmol amino group) and pyridine (1.5 mmol) were added under nitrogen atmosphere. The mixture was stirred at 40 °C for 16 h and thereafter filtered and washed with HCl, K2CO3, distilled water, ethanol and large amounts of CH2Cl2 in order to obtain IPhCONH-propyl silica gel. FTIR (KBr): ν = 3423 (m, νN–H), 2939 (s, νN–H), 1649 (s, νCO), 1099 (s, νSi–O–Si) cm−1.
For the three-phase test, a 50 mL Schlenk flask was charged with 1 g of IPhCONH-propyl silica gel, 2-iodoacetophenone (1 mmol), phenylboronic acid (2.2 mmol), [Pd(1-BI)2Cl2] (0.01 mmol) and Na2CO3 (3 mmol). Then, 5 mL of distilled water was added. The Schlenk flask was evacuated and filled with CO. Under the balloon pressure of CO, the reaction mixture was stirred at 80 °C for 4 h. After that time, the Schlenk flask was cooled down, and the organic products were extracted with 3 × 7 mL of n-hexane. The extract was GC analyzed with mesitylene as the internal standard (0.076 mL, 5.46 × 10−4 mol). The solid was separated by filtration washed with ethanol and CH2Cl2. The solid was hydrolyzed at 90 °C for 3 days with KOH in ethanol/water (3.4 g KOH in 20 mL of EtOH plus 10 mL of H2O). The resulting mixture was neutralized with aqueous HCl and extracted first with n-hexane, then with dichloromethane. The extracts were analyzed by GC.
(3) Alkoxycarbonylation reaction
A 50 mL Schlenk flask was charged with aryl halide (1 mmol), [Pd(1-BI)2Cl2] (0.01 mmol), and NEt(i-Pr)2 (3 mmol). Then, 5 mL of ROH:H2O (1:1) was added. The Schlenk flask was evacuated and filled with CO. Under the balloon pressure of CO, the reaction mixture was stirred at 60 °C for 2 h. After that time, the Schlenk flask was cooled down, and the organic products were extracted with 3 × 7 mL of n-hexane (3 × 15 min with stirring). The extract was GC analyzed with mesitylene as the internal standard (0.076 mL, 5.46 × 10−4 mol). After the solvents were evaporated, the crude product was purified by column chromatography on silica gel using hexane/ethyl acetate (10:1) as the eluent, the corresponding ester was obtained.
Results and discussion
The reduction of Pd(im)2Cl2 complexes to Pd NPs was examined at RT under 1 atm of CO in water. Under these conditions, without any additional reducing agent, a black precipitate was formed already after 5 min. The stirring of the reaction mixture was continued, and, after 1 h, Pd NPs with the average diameter of 2–3 mm were detected by TEM (Fig. 1). Interestingly, Pd NPs remained non-agglomerated although no additional stabilizer was added. | ||
| Fig. 1 TEM micrograph and Pd NPs size distribution. | ||
The coupling of iodobenzene (PhI) and phenylboronic acid (PhB(OH)2) under CO atmosphere was selected as a model reaction to study (Scheme 1). Product 1, diphenyl ketone, was expected as the desired one, whereas biphenyl (2) can be formed as a product of the classic Suzuki coupling without CO insertion.
 | ||
| Scheme 1 Carbonylative Suzuki coupling of iodobenzene with phenylboronic acid. | ||
When Pd(1-BI)2Cl2 was used as a catalyst precursor, 89% of benzophenone was obtained. The aim of further experiments was to examine the activity of the pre-formed Pd NPs (Fig. 2). Thus, Pd(1-BI)2Cl2 was treated with CO in water for 10 min or for 3 h before the introduction of substrates. Next, the reaction mixture was stirred for 1 h at 80 °C. In both experiments, despite the different pre-treatment time, very similar results were obtained; namely, the conversion was 84% with selectivity to benzophenone being 83% (Table 1). In addition, the results were close to those obtained for the precursor used without any pre-treatment, suggesting the fast formation of Pd NPs in the catalytic conditions. Only a slightly lower conversion was noted at a lower amount of Pd (0.5% mol). Also, in this case, the catalytic results were not affected by the catalyst pre-treatment time (Table 1).
 | ||
| Fig. 2 XRD picture of Pd NPs. | ||
| Pd precursor | Pre-treatment time (min) RT, 1 atm CO | Conversion (%) | 1 (%) | Selectivity to 1 (%) |
|---|---|---|---|---|
| a Reaction conditions: PhI (1 mmol), PhB(OH)2 (1.2 mmol), CO (1 atm), Na2CO3 (3 mmol), water (5 mL), 80 °C, 1 h. | ||||
| Pd(1-BI)2Cl2, 1 mol% | — | 99 | 89 | 90 |
| Pd(1-BI)2Cl2, 1 mol% | 10 | 88 | 73 | 83 |
| Pd(1-BI)2Cl2, 1 mol% | 180 | 84 | 70 | 83 |
| Pd(1-BI)2Cl2, 0.5 mol% | 10 | 76 | 57 | 75 |
| Pd(1-BI)2Cl2, 0.5 mol% | 180 | 79 | 58 | 74 |
| PdCl2, 1 mol% | — | 36 | 14 | 38 |
| PdCl2, 1 mol% | 180 | 26 | 18 | 68 |
| PdCl2, 1 mol% + 1-BI, 2 mol% | — | 56 | 41 | 72 |
A different situation was observed for PdCl2 used as the source of Pd NPs. The conversion was only 36%, and it decreased to 26% when PdCl2 was treated with CO for 3 h before the addition of the substrates. It can be assumed that Pd NPs formed from PdCl2 undergo much faster deactivation than those originating from Pd(im)2Cl2 precursors (Table 1). However, the catalytic activity of PdCl2 was significantly improved by addition of free imidazole (1-BI) (Table 1). Thus, conversion increased to 56% and 41% of coupling product 1 was formed confirming an important role of imidazole in the stabilization of Pd NPs.
In order to evidence the catalytic role of Pd NPs in the studied reaction, the Hg(0) test was done.31–34 When Hg(0) was introduced at the beginning of the reaction, the conversion of iodobenzene was 12%, significantly less than in the reference experiment performed without Hg(0), which gave a 99% conversion (Table 2). In the second test, Hg(0) was added after 30 min, and, in this case, the reaction stopped at 47% conversion. These experiments confirmed that Pd NPs were involved in the catalytic process because their elimination by amalgam formation inhibited the reaction course.31–34 Interestingly, our system differed from two other systems containing Pd NPs, in which soluble Pd species were responsible for the catalytic activity.26,27
| Conversion (%) | 1 (%) | 2 (%) | |
|---|---|---|---|
| a Reaction conditions: 80 °C, [Pd] 1 mol%, PhI (1 mmol), PhB(OH)2 (1.2 mmol), CO (1 atm), Na2CO3 (3 mmol), water 5 mL, 1 h, [Hg]/[Pd] 200. | |||
| Without Hg | 99 | 89 | 10 |
| Hg added at the beginning of the reaction | 12 | 8 | 4 |
| Hg added after 30 min | 47 | 34 | 13 |
| CS2, 0.65 mol% | 77 | 72 | 5 |
| CS2, 1 mol% | 53 | 52 | 1 |
Addition of CS2 to the reaction mixture also resulted in decrease of both, conversion of PhI and yield of product 1. As expected, the poisoning effect caused by blocking of the surface of Pd NPs limiting access of substrates, was stronger at higher amount of CS2 (1 mol%) (Table 2).32,34
Surprisingly, even stronger inhibition of the catalytic reaction was noted in the presence of TBAB. Conversion decreased to 32% or 27% after addition of 20-fold or 40-fold excess of TBAB in relation to palladium. Although increased stabilization of Pd NPs was expected after introduction of TBAB, another effect should be considered, which comprises dissolving of Pd NPs and formation of less active soluble Pd species. Dissolution of Pd NPs was earlier evidenced in their reaction with PhI in the presence of TBAB.35 However in contrast to the carbonylative Suzuki coupling, in Heck reaction formation of soluble Pd species improved significantly the reaction yield.35
The three-phase test, a useful method of identification of catalytically active homogeneous palladium species,32 was performed according to the described procedure.36 Thus, 2-iodobenzyl chloride was immobilized on aminopropyl functionalized silica gel. The carbonylative Suzuki reaction was then carried out with the supported reagent and with 2-iodoacetophenone as a free available aryl halide. According to the CG analysis, 2-iodoacetophenone did not formed carbonylative coupling product, however formation of classic Suzuki product was evidenced. Thereafter, the amide was hydrolyzed from the support. The mixture was neutralized and extracted first with n-hexane, then with dichloromethane. The extracts were analyzed by GC. Formation of 2-benzoyl benzoic acid and 2-diphenylcarboxylic acid did not take place in agreement with the suggested reaction pathway.
The results of the optimization of the reaction conditions with respect to temperature and time are presented in Table 3. Considering the effect of the imidazole ligand on the catalytic reaction course, Pd(1-BI)2Cl2 afforded slightly higher conversions, and it was selected for further reactions. Because conversion increased as the temperature increases, we decided to perform further catalytic tests at 80 °C in 1 h.
| Pd precursor | Temp. (°C) | Time (h) | Conv.b (%) | 1 b (%) | 2 b (%) | Selectivity to 1 (%) |
|---|---|---|---|---|---|---|
| a [Pd] (1 mol%), Na2CO3 (3 mmol), water (5 mL), PhI (1 mmol), PhB(OH)2 (1.2 mmol), CO (1 atm). b Conversions were determined by GC using mesitylene as an internal standard. | ||||||
| Pd(1-MI)2Cl2 | 60 | 2 | 68 | 65 | 3 | 96 |
| Pd(1-MI)2Cl2 | 60 | 4 | 91 | 87 | 4 | 96 |
| Pd(1-MI)2Cl2 | 80 | 2 | 95 | 89 | 6 | 94 |
| Pd(1-MI)2Cl2 | 80 | 1 | 87 | 77 | 10 | 89 |
| Pd(1-BI)2Cl2 | 80 | 2 | 100 | 93 | 7 | 93 |
| Pd(1-BI)2Cl2 | 80 | 1 | 99 | 89 | 10 | 90 |
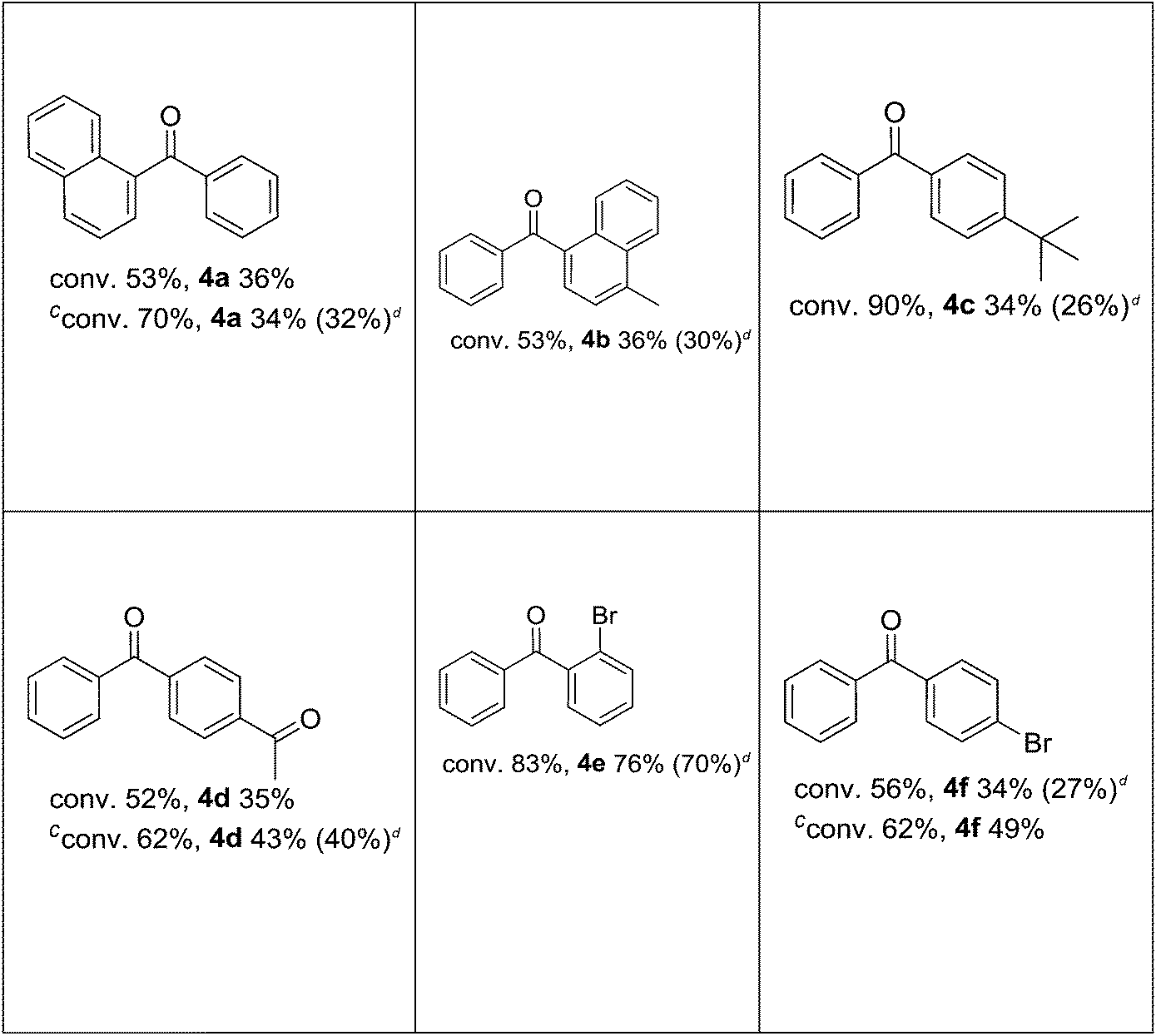
After defining the optimal conditions, it was investigated how substituted iodobenzenes affected the reaction. As shown in Table 4, the palladium-catalyzed carbonylative Suzuki reaction proceeded efficiently for these substrates under low pressure (1 atm CO, 80 °C) in water (Table 4). Ortho-, meta-, and para-substituted methoxy-iodobenzenes gave the corresponding diaryl ketones in moderate to good yields (Table 4). As seen in Table 4, a good yield and an excellent selectivity were achieved for diaryl ketones with iodobenzene derivatives bearing electron donating groups. Then again, a decrease in selectivity was detected in reactions carried out with an iodobenzene derivative bearing a para-substituted electron withdrawing group (Table 4).


In reaction with 2-iodobenzyl alcohol (Table 4), intramolecular hydroxycarbonylation took place instead of the carbonylative coupling. As a result, 1-phthalanone was formed as the only product. A similar result (81% of 1-phthalanone) was also obtained in dioxane under the carbonylative Suzuki conditions.
The efficient procedure of isolation of ketone from the reaction mixture was also elaborated (see the Experimental part), which resulted in obtaining of 85% of pure benzophenone.
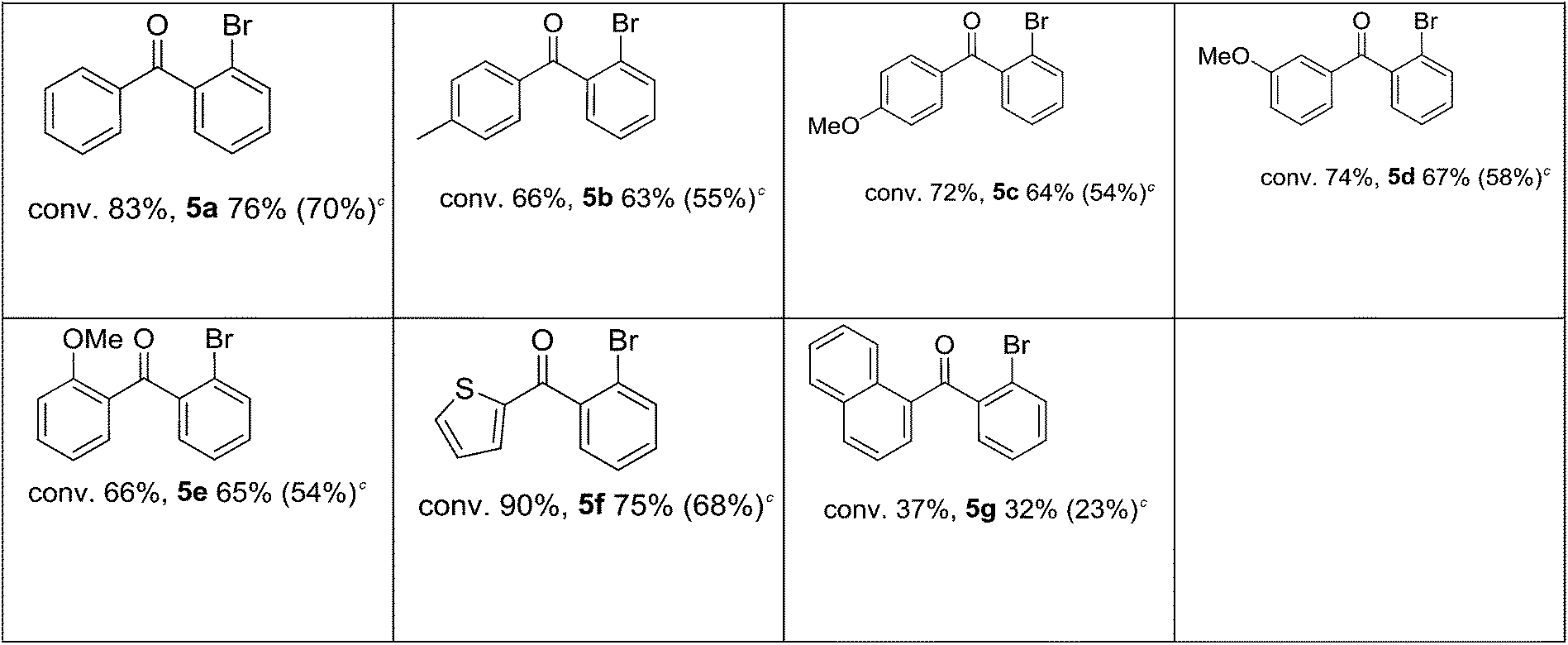
After the influence of substituted iodobenzenes on the studied reaction was recognized, the effect of boronic acid derivatives was investigated (Table 5). For the desired coupling products, both the yield and the selectivity stayed at a moderate level in the test reactions implemented with phenylboronic acid derivatives bearing a para-substituted electron withdrawing and electron donating group (Table 5). Attempts to improve the reaction efficiency by extending the reaction time gave positive results only for 4-bromophenylboronic acid. In that case, both the yield of ketone and the selectivity increased. Conversely, reactions of 4-acylphenylboronic acid and naphtylboronic acid over a longer time produced mainly biaryls, and, as a result, selectivity to the carbonylation product decreased (Table 5). The best results were obtained for 2-bromophenylboronic acid, which formed 76% of ketone after 1 h.

Test reactions were then performed for different iodobenzenes and 2-bromophenylboronic acid (Table 6). It was established that the reactions gave diaryl ketones with a good yield and a very high selectivity (83–99%) for all the tested iodobenzenes (Table 6).

The possibility of double carbonylation with in situ generated Pd NPs was studied for 1-bromo-4-iodobenzene (Scheme 2).
 | ||
| Scheme 2 Carbonylative Suzuki coupling of 1-bromo-4-iodobenzene. | ||
A mixture of products was obtained, and, importantly, the formation of products II and III indicated that not only a C–I but also a C–Br bond was activated in the studied system (Table 7). The highest yield of product II, 9%, was obtained at the presence of Na2CO3. In contrast, amine N(i-Pr)2Et promoted formation of Suzuki products. In this case activation of C–Br bond was the most efficient and consequently high yield of III (21%) and V (15%) was noted.
| Temp. [°C] | Base | Time [h] | Conv.b [%] | I | II | III | IV | V |
|---|---|---|---|---|---|---|---|---|
| a 1-Bromo-4-iodobenzene (1 mmol), PhB(OH)2 (2.2 mmol), Pd(1-BI)2Cl2 (1 mol%), Na2CO3 (3 mmol), CO (1 atm.), water (5 mL), 80 °C, 1 h. b Conversions were determined by GC using mesitylene as internal standard. | ||||||||
| 80 | Na2CO3 | 1 | 54 | 31 | 9 | 3 | 10 | 1 |
| 80 | Na2CO3 | 4 | 75 | 45 | 4 | 7 | 18 | 1 |
| 80 | Na2CO3 | 5 | 79 | 46 | 6 | 8 | 17 | 2 |
| 80 | N(i-Pr)2Et | 1 | 96 | 4 | 6 | 21 | 50 | 15 |
| 80 | KOH | 1 | 64 | 29 | 7 | 6 | 18 | 4 |
| 100 | Na2CO3 | 1 | 74 | 18 | 9 | 15 | 28 | 4 |
At elevated pressure, 5 or 10 bar CO, 1-bromo-4-iodobenzene was converted to product I with high selectivity (Table 8).
Surprisingly, conversion of 1,4-diiodobenzene at 1 bar of CO reached only 45% with formation of III as a main product (Scheme 3). However selectivity to dicarbonylation product II increased significantly after increase of pressure to 10 bar. The best results were obtained after 18 h reaction (Table 9).
 | ||
| Scheme 3 Carbonylative Suzuki coupling of 1,4-diiodobenzene. | ||
| Pressure of CO | Time [h] | Conv.b [%] | I | II | III | IV | V |
|---|---|---|---|---|---|---|---|
| a 1,4-Diiodobenzene (1 mmol), PhB(OH)2 (2.2 mmol), Pd(1-BI)2Cl2 (1 mol%), Na2CO3 (3 mmol), water (5 mL), 80 °C, 1 h. b Conversions were determined by GC using mesitylene as internal standard. | |||||||
| 1 | 1 | 16 | 1 | 4 | 4 | 1 | 6 |
| 1 | 1 | 19 | 0 | 2 | 7 | 2 | 8 |
| 1 | 5 | 41 | 1 | 10 | 25 | 1 | 4 |
| 1 | 5 | 45 | 2 | 9 | 25 | 2 | 7 |
| 10 | 5 | 50 | 2 | 46 | 0 | 0 | 2 |
| 10 | 18 | 66 | 1 | 54 | 0 | 0 | 11 |
The same catalytic system was also tested in other solvents such as IPA:water (1:1) (IPA = 2-propanol) and EtOH (Table S1†).
In the IPA:water (1:1) system, the selectivity remained low, and it changed depending on the base from 26 to 71%. For example, a high conversion (96%) was obtained with the KOH base; however, only 25% of 1 was formed. High conversions were also attained in the IPA:water (1:1) system with organic bases, NEt3 and NEt(i-Pr)2, but even then good selectivity was not achieved due to the formation of isopropylbenzoate, an alkoxycarbonylation product (Scheme 4).
 | ||
| Scheme 4 Effect of solvent and base on carbonylative Suzuki coupling. | ||
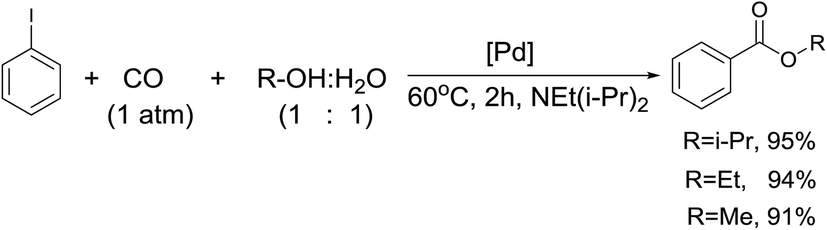
Finding benzoate esters in the reaction products prompted us to study the catalytic activity of our system in alkoxycarbonylation. Accordingly, the catalytic reaction was performed without PhB(OH)2 in ROH:water (1:1). The corresponding esters were formed as the only products with a yield of 91–95% (isolated yield was equal 80–87%) (Scheme 5).
 | ||
| Scheme 5 Formation of benzoate esters via alkoxycarbonylation. | ||
In order to check whether Pd NPs were responsible for alkoxycarbonylation, the Pd precursor was treated with CO in IPA:water and an as-prepared solution was next used for the catalytic reaction (Scheme 6).
 | ||
| Scheme 6 The effect of the pre-treatment time on the catalytic activity of Pd NPs in alkoxycarbonylation. | ||
In both reactions, the ester was obtained with high yields confirming the ability of Pd NPs formed in situ to catalyze alkoxycarbonylation. Interestingly, Pd NPs formed from Pd–imidazole complexes remained active without any additional agents in contrast to the fast deactivation of Pd NPs obtained from PdCl2(cod).37 When PdCl2(cod) was used in the methoxycarbonylation of iodobenzene, the conversion to ester was in the order of 20%, and an increase in conversion was only achieved in the presence of salts, such as [Bu4N]X,36 or ionic liquids.38,39
Conclusions
Functionalized diaryl ketones were obtained in moderate to very good yields in water using Pd NPs generated in situ from palladium complexes with imidazole ligands. Pd NPs formed from Pd–imidazole precursors in water under CO atmosphere were stable and non-agglomerated, probably as a result of the stabilizing effect of imidazoles. The elaborated carbonylative coupling procedure, based on a one-step synthesis, can be successfully applied for the preparation of arylated ketones from different iodobenzenes and aryl boronic acid derivatives. High selectivity to desired carbonylation products is the important advantage of this catalytic procedure. The same catalytic system used for the alkoxycarbonylation of iodobenzene produced an ester with high efficiency. In reactions with 1-bromo-4-iodobenzene both C–X bonds (C–I and C–Br) were activated to form coupling products.Acknowledgements
Financial support of National Science Centre (NCN, Poland) with grant 2014/15/B/ST5/02101 (P. W., A. M. T.) is gratefully acknowledged.References
- S. Budavari, The Merck Index, Merck, Rahway, USA, 11th edn, 1989 Search PubMed.
- R. K. Dieter, Tetrahedron, 1999, 55, 4177–4236 CrossRef CAS.
- R. Skoda-Földes, Z. Székvölgyi, L. Kollár, Z. Berente, J. Horváth and Z. Tuba, Tetrahedron, 2000, 56, 3415–3418 CrossRef.
- G. A. Olah, Friedel–Crafts Chemistry, Wiley, New York, 1973 Search PubMed.
- A. Fürstner, D. Voigtländer, W. Schrader, D. Giebel and M. T. Reetz, Org. Lett., 2001, 3, 417–420 CrossRef.
- E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316–1327 CrossRef CAS PubMed.
- G. F. Silbestri, R. Bogel-Masson, M. T. Lockhart and A. B. Chopa, J. Organomet. Chem., 2006, 691, 1520–1524 CrossRef CAS.
- W. P. Neumann, H. Hillgärtner, K. M. Baines, R. Dicke, K. Vorspohl, U. Kobs and U. Nussbeutel, Tetrahedron, 1989, 45, 951–960 CrossRef CAS.
- J. W. Labadie and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 6129–6137 CrossRef CAS.
- L. J. Gooβen and K. Ghosh, Angew. Chem., Int. Ed., 2001, 40, 3458–3460 CrossRef.
- H. Tatamidani, F. Kakiuchi and N. Chatani, Org. Lett., 2004, 6, 3597–3599 CrossRef CAS PubMed.
- M. K. Eberle and G. G. Kahle, Tetrahedron Lett., 1980, 21, 2303–2304 CrossRef CAS.
- J.-J. Brunet and R. Chauvin, Chem. Soc. Rev., 1995, 24, 89–95 RSC.
- S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367–10389 RSC.
- B. M. O'Keefe, N. Simmons and S. F. Martin, Org. Lett., 2008, 10(22), 5301–5304 CrossRef PubMed.
- S. Couve-Bonnaire, J.-F. Carpentier, A. Mortreux and Y. Castanet, Tetrahedron, 2003, 59, 2793–2799 CrossRef CAS.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC.
- M. B. Andrus, Y. Ma, Y. Zang and C. Song, Tetrahedron Lett., 2002, 43, 9137–9140 CrossRef CAS.
- K. M. Bjerglund, T. Skydstrup and G. A. Molander, Org. Lett., 2014, 16, 1888–1891 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Tetrahedron Lett., 2010, 51, 6146–6149 CrossRef CAS.
- X.-F. Wu, H. Neumann and M. Beller, Adv. Synth. Catal., 2011, 353, 788–792 CrossRef CAS.
- H. Li, M. Yang, Y. Qi and J. Xue, Eur. J. Org. Chem., 2011, 2662–2667 CrossRef.
- P. J. Tambade, Y. P. Patil, A. G. Panda and B. M. Bhanage, Eur. J. Org. Chem., 2009, 3022–3025 CrossRef CAS.
- S.-Z. Zheng, L.-W. Xu and C.-G. Xia, Appl. Organomet. Chem., 2007, 21, 772–776 CrossRef CAS.
- H. Zhao, G. Zheng, S. Sheng and M. Cai, Catal. Commun., 2009, 11, 158–161 CrossRef CAS.
- N. Jiao, Z. Li, Y. Wang, J. Liu and C. Xia, RSC Adv., 2015, 5, 26913–26922 RSC.
- Q. Zhou, S. Wei and W. Han, J. Org. Chem., 2014, 79, 1454–1460 CrossRef CAS PubMed.
- C.-J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed.
- M. S. Szulmanowicz, W. Zawartka, A. Gniewek and A. M. Trzeciak, Inorg. Chim. Acta, 2010, 363, 4346–4354 CrossRef CAS.
- H. Gruber-Woelfler, P. F. Radaschitz, P. W. Feenstra, W. Haas and J. G. Khinast, J. Catal., 2012, 286, 30–40 CrossRef CAS PubMed.
- F. Fernandez, B. Cordero, J. Durand, G. Muller, F. Malbose, Y. Kihn, E. Teuma and M. Gomez, Dalton Trans., 2007, 5572–5581 RSC.
- N. T. S. Phan, M. van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- S. Perdiau, S. Harder, H. J. Heeres and J. G. de Vries, ChemSusChem, 2012, 5, 2427–2434 CrossRef PubMed.
- J. A. Widergen and R. F. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef.
- A. Gniewek, A. M. Trzeciak, J. J. Ziółkowski, L. Kępiński, J. Wrzyszcz and W. Tylus, J. Catal., 2005, 229, 332–343 CrossRef CAS.
- H. Gruber-Woelfler, P. F. Radaschitz, P. W. Feenstra, W. Haas and J. G. Khinast, J. Catal., 2012, 286, 30–40 CrossRef CAS PubMed.
- A. M. Trzeciak, W. Wojtków, J. J. Ziółkowski, J. Wrzyszcz and M. Zawadzki, New J. Chem., 2004, 28, 859–863 RSC.
- W. Wojtków, A. M. Trzeciak, R. Choukroun and J. L. Pellegatta, J. Mol. Catal. A: Chem., 2004, 224, 81–86 CrossRef.
- W. Zawartka, A. M. Trzeciak, J. J. Ziółkowski, T. Lis, Z. Ciunik and J. Pernak, Adv. Synth. Catal., 2006, 348, 1689–1698 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02889f |
| This journal is © The Royal Society of Chemistry 2016 |