
Formation of a robust Ru4O4 skeleton with two Ru2(CO)4 units in criss–cross configuration†
Abstract
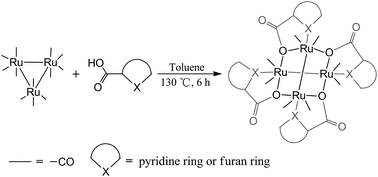
Thermolysis of triruthenium dodecacarbonyl Ru3(CO)12 with picolinic acid, 2-furoic acid and 2-thiophenecarboxylic acid was thoroughly studied. Three ruthenium carbonyl products, Ru4(CO)8(μ2-O, η1-N-pic)4 (1) (pic = picolinate), [Ru2(CO)4(fur)2]m (2) (fur = 2-furoate) and [Ru2(CO)4(thi)2]n (3) (thi = 2-thiophenecarboxylate) were isolated. Single crystal X-ray crystallography revealed that compound 1 is a tetraruthenium cluster with two Ru2(CO)4 units in an unexpected criss–cross geometry, while compounds 2 and 3 exhibit the classical sawhorse structure. Compound 1 was stable to common donor ligands and formed solvated compounds Ru4(CO)8(μ2-O, η1-N-pic)4·H2O (4) and Ru4(CO)8(μ2-O, η1-N-pic)4·CH3CN (5) in hydrous toluene and acetonitrile, respectively; compounds 2 and 3 converted into monomers Ru2(CO)4(fur)2(H2O)2·H2O (2b) and Ru2(CO)4(thi)2(CH3OH)2·CH3OH (3b) in hydrous dichloromethane and acetonitrile, respectively. [Ru2(CO)4(pic)2]2 (1a), Ru2(fur)2(CO)6 (2a) and Ru2(thi)2(CO)6 (3a) were proposed as the corresponding intermediates of 1–3 based on in situ FT-IR spectroscopy, LC-MS and the molecular structures of the known ruthenium carbonyl carboxylates.
 Please wait while we load your content...
Please wait while we load your content...

