
Enhanced electrocatalytic activity of three-dimensionally-ordered macroporous La0.6Sr0.4CoO3−δ perovskite oxide for Li–O2 battery application†
Mi Young
Oh
a,
Jong Ju
Lee
b,
Awan
Zahoor
d,
G.
Gnana kumar
e and
Kee Suk
Nahm
*abc
aR&D Education Center for Fuel Cell Materials & Systems, Republic of Korea. E-mail: nahmks@jbnu.ac.kr; Fax: +82 63 270 3909; Tel: +82 63 270 2311
bDepartment of Energy Storage and Conversion Engineering, Republic of Korea
cSchool of Chemical Engineering, Chonbuk National University, Jeonju 561-756, Republic of Korea
dDepartment of Chemical Engineering, N.E.D. University of Engineering & Technology, University Road, Karachi, 75270, Pakistan
eDepartment of Physical Chemistry, Madurai-Kamaraj University, Madurai-625 021, India
First published on 21st March 2016
Abstract
Three-dimensionally-ordered macroporous perovskite La0.6Sr0.4CoO3−δ (3DOM LSC) structures were synthesized using polymethyl methacrylate (PMMA) templates prepared with different techniques. The electrocatalytic activity of the prepared 3DOM LSC perovskites was examined for both the oxygen reaction (ORR) and oxygen evolution reaction (OER) using various electrochemical techniques in both aqueous and nonaqueous electrolyte solutions. The discharge capacity and cycle performance were also investigated with a lithium–air battery fabricated with the 3DOM LSC perovskite catalysts. It was found that the electrochemical study in both aqueous and nonaqueous electrolyte solutions and the performance measurement in the lithium–air battery exhibit a similar electrocatalytic activity in the order of Centrifuge LSC1000 > Pellet LSC1000 > Powder LSC1000. The best performance of a lithium–air battery is observed from Centrifuge LSC1000. The enhanced catalytic performance of the 3DOM structures could be reasonably attributed to the formation of porous structures with increased surface area and catalytic activity, which facilitate the fast reversible formation and decomposition of Li2O2 as well as quick transportation of lithium ions and oxygen in the lithium air cell.
1. Introduction
The lithium–air battery has been reported to generate a much higher theoretical energy density than any other secondary batteries and the oxygen in air could be used as the active material in air cathode.1–6 However, the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) that takes place in the battery are still complicated with multi-electron transfer, resulting in the sluggish kinetics. In addition, the solid-phase reaction products such as lithium peroxide (Li2O2) that precipitate at the cathode during discharge do not recover completely during recharge which leads to the low efficiency of the battery.7–9 At this context catalyst is vital to smoothen the battery processes and improve the kinetics. An advanced bifunctional catalyst is required to decrease the overpotential and consequently increase the electrochemical efficiency of the battery.10,11Recently perovskite oxides have emerged as efficient cathode catalysts because of the chemically stable structure, mixed electronic/ionic conductivity by composition, and oxidation/reduction catalytic activities. The perovskite oxides have been reported to exhibit good cation ordering and provide disorder-free channels of oxygen vacancies to enhance the mobility of oxygen ions that would benefits the ORR and OER reactions.12,13 So far the catalytic performance of various perovskite oxides such as CaMnO3,13 LaxSr1−xCoO3,14–16 LaxSr1−xMnO3,17,18 La2NiO4,19 La0.6Ca0.4Co0.8Fe0.2O3, La0.6Sr0.4Co0.2Fe0.8O3,20 La0.6Sr0.4Fe0.6Mn0.4O3,21 Ba0.9Co0.5Fe0.4Nb0.1O3,22 LaFeO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and LaNi1−xMgxO324 had been investigated as active air cathodes catalyst for lithium–air battery applications. However, owing to the lower specific surface area most of the previously reported perovskite oxides samples have not achieved their maximum electrocatalytic activity in lithium–air battery. Hence it is necessary to examine and understand the structural design of perovskite oxides to improve their surface area and thereby the catalytic performances.
23 and LaNi1−xMgxO324 had been investigated as active air cathodes catalyst for lithium–air battery applications. However, owing to the lower specific surface area most of the previously reported perovskite oxides samples have not achieved their maximum electrocatalytic activity in lithium–air battery. Hence it is necessary to examine and understand the structural design of perovskite oxides to improve their surface area and thereby the catalytic performances.
Ball milling and micro- or nano-structure fabrication techniques had been an effective method to increase the active surface area of catalysts which would consecutively enhance the catalytic activity of the catalyst.25–27 The crystallite size and crystallinity generated during the ball milling process had been proven to be in strong correlation with the enhancement of electrochemical activity of the materials.28 For example, microporous La0.8Sr0.2MnO3 nanorods28 and three dimensional ordered macroporous LaFeO329 provided much higher surface areas with numerous defects than their powdered samples, that was beneficial to the ORR and OER in the discharge and charge processes, respectively.
In the previous work,14 La0.6Sr0.4CoO3−δ (LSC) perovskite as air cathode catalyst of lithium–air batteries was extensively studied. LSC was utilized because of their highest electronic and ionic conductivities among the reported perovskite catalysts.30–33 Even though the LSC exhibited superior catalytic property in oxygen evolution reaction, lower battery performance was observed. Nian Sun et al.16 also studied the La0.6Sr0.4CoO3−δ perovskite oxide which was synthesized by a dry gel combustion. They reported that the initial discharge capacity of LSC was 1237.5 mA h g−1 at the current densities of 80 mA g−1 and the discharge capacity was maintained 16 times when the cut-off capacity was set as 500 mA h g−1, which are much worse than our report.14 It was also conclusive that ball milling increased the surface area of LSC thereby effectively enhancing the performance of lithium air battery. So in order to increase the surface area, and improve the battery performance, we have attempted to synthesize 3D-ordered macroporous perovskite La0.6Sr0.4CoO3−δ (3DOM LSC) structures.
In this work, 3D-ordered macroporous perovskite La0.6Sr0.4CoO3−δ (3DOM LSC) structures were synthesized using a template of polymethyl methacrylate (PMMA) spheres. The PMMA template was fabricated by two methods, a 13 mm diameter pellet type array of PMMA spheres under pressure and a close-pack PMMA spheres (2 g) into colloidal crystals by centrifugation. The structure of the synthesized 3DOM LSC was characterized using various analytic techniques. The electrocatalytic activities of the 3DOM LSC catalysts for ORR and OER were systematically studied in both aqueous and non-aqueous electrolyte solutions using various electrochemical analytic measurements and were compared with that of conventional Powder LSCs. The battery performance of a lithium air battery fabricated with thus prepared perovskite structures was also evaluated using a custom-built Swagelok™ type test cell. On the basis of the experimental results, we have intensively discussed the superiority of the prepared 3DOM LSC structure as suitable bifunctional catalyst of lithium–air battery.
2. Experimental
2.1 Preparation of 3DOM structure La0.6Sr0.4CoO3−δ
The 3DOM La0.6Sr0.4CoO3−δ structure with nanoporous walls was synthesized using a colloidal crystal templating process (CCT).34,35 The CCT has been proposed to synthesize 3D-ordered macroporous materials with pores sized in the micro or nanometer range for applications in filters, sensors, fuel cells, catalysis, and lithium ion batteries.35–40 The synthetic process is as follows. Organic polymer templates are first fabricated via self-assembly using gravitational sedimentation, centrifugation, or filtration methods, and then metal oxide precursors fill the empty spaces in the template. Finally, 3D-ordered metal oxide structures are produced after removing the template through calcinations or extraction.For the preparation of La0.6Sr0.4CoO3−δ precursor solution, stoichiometric amounts of La(NO3)3·6H2O (99.9%, Sigma-Aldrich), Sr(NO3)2 (99%, Sigma-Aldrich), Co(NO3)2·6H2O (97.7%, Sigma-Aldrich) [La(NO3)3·6H2O:Sr(NO3)2:Co(NO3)2·6H2O = 0.6:0.4:1] were dissolved in ethylene glycol (99%, Sigma-Aldrich) solution used as an esterification agent with stirring for two hours. To increase the solubility of La, Sr, and Co nitride salts, methanol anhydrous (99.8%, Sigma-Aldrich) was added to the mixed solution and stirred for another hour. Also, methanol anhydrous ensured that the PMMA template was adequately wetted with the mixed precursor solution by decreasing the viscosity of ethylene glycol. The PMMA template was fabricated in two ways in this experiment, as shown in a schematic illustration in Fig. S1.† The first way was creation of a 13 mm-diameter pellet-type array through application of 4 tons of pressure on 2 g of PMMA spheres (with an average diameter of ca. 5 ± 3 μm) using an evacuable Pellet Press (PIKE Technologies, PN161-1900). The second way was to close-pack PMMA spheres (2 g) into colloidal crystals by centrifugation (Hanil Science, Combi-514R) (4000 rpm, three times for 20 min each). The former method is called Pellet LSC in the present study, while the latter is Centrifuge LSC. Then, thus prepared PMMA templates were soaked in the metal precursor solution for 2 hours in order to fully infiltrate the solution into the unfilled spaces in the templates. After soaking in the precursor solution, the remaining solution was filtered through a Büchner funnel, and the solid residues were dried at room temperature for 24 hours. This dried solid was calcined in air to remove PMMA spheres. The calcination process was performed in a box furnace (AJ-S84) for 5 hours at two different temperatures (650 °C and 1000 °C) at a heating rate of 11 °C min−1. The prepared final products are called Pellet LSC650, Pellet LSC1000, Centrifuge LSC650, and Centrifuge LSC1000 depending on the template alignment method and calcination temperature. For comparison, powder-type La0.6Sr0.4CoO3−δ was also synthesized at 1000 °C using the Pechini method41,42 and is called LSC1000 in this paper.
2.2 Structural characterization of synthesized samples
X-ray diffraction (XRD) with a CuKα (λ = 1.54059 Å) target in the 2θ range of 10–100° was used to analyze the crystal structure of the synthesized 3DOM La0.6Sr0.4CoO3−δ catalysts. A field emission scanning electron microscopy (FESEM) was used to measure particle size, distribution, and morphology. Also, the Brunauer–Emmett–Teller (BET) technique was employed to characterize the specific surface area of the synthesized La0.6Sr0.4CoO3−δ catalysts.2.3 Electrochemical characterization of synthesized samples
Electrocatalytic characteristics of the synthesized 3DOM La0.6Sr0.4CoO3−δ catalysts were examined using electrochemical analytic techniques such as cyclic voltammetry (CV) and linear sweep voltammetry (LSV) for ORR and OER studies. For CV, ORR, and OER studies, the synthesized LSC was mixed with carbon powder (Cabot Vulcan XC-72) at a weight ratio of 3:7 to ensure sufficient electronic conductivity. Ten milligrams of as-prepared catalyst was dispersed ultrasonically in 150 μL of diluted Nafion alcohol solution (5 wt%) dissolved in isopropyl alcohol (IPA), and about 13.5 μL of the suspension was pipetted onto a glassy carbon substrate. Platinum wire and Hg/HgO were used as the counter and reference electrodes, respectively. Prior to measurement, O2 was bubbled directly into the cell for at least one hour. The detailed fabrication process of the electrode used for the electrochemical test was reported in our previous study.14
2.4 Performance evaluation of a lithium–air battery
The electrochemical performance of the Li–O2 battery was measured using a custom-built Swagelok test cell. The air cathodes were prepared by mixing the synthesized 3DOM LSC catalyst, KB conductive carbon, and teflonized acetylene black (TAB) binder (60%) in isopropyl alcohol (IPA). The mixing ratio of the 3DOM LSC catalyst and KB conductive carbon was 1:2. The prepared electrode was dried in a vacuum at 100 °C for 12 hours. All cells were assembled in a glove box under an Ar atmosphere, using a lithium metal foil anode, glass fiber separator, oxygen cathode, and electrolyte containing 1 M LITFSI in TEGDME. The cell test was performed under oxygen flow (10 cm3 min−1) at a current density of 0.1 mA cm−2 in a cut-off voltage range of 2.0–4.3 V.
3. Results and discussion
3.1 Structure analysis
Fig. 1 shows XRD patterns of the synthesized 3DOM La0.6Sr0.4CoO3−δ samples for different synthetic processes and conditions. Powder-type La0.6Sr0.4CoO3−δ (Powder LSC1000) was also characterized using XRD for comparison, as shown in Fig. 1(e). It can be seen from the XRD spectra that all the samples are synthesized with the structure of perovskite La0.6Sr0.4CoO3−δ. All the XRD spectra show a main characteristic peak at 2θ = 33° corresponding to the (110)/(104) plane of La0.6Sr0.4CoO3−δ, and the peaks observed from the spectra coincide well with the previously reported XRD spectrum of La0.6Sr0.4CoO3−δ.14 For 3DOM LSCs synthesized at a calcination temperature of 650 °C, however, the SrCO3 and Co3O4 impurity phases are clearly observed in both Pellet LSC650 and Centrifuge LSC650. On the other hand, when the LSC samples were synthesized at a calcination temperature of 1000 °C, XRD measurements revealed a stable and pure perovskite La0.6Sr0.4CoO3−δ structure with no impurities, as discussed in our previous report.14 | ||
| Fig. 1 XRD patterns of 3DOM La0.6Sr0.4CoO3−δ perovskites. | ||
Primary particle sizes of the as-prepared LSC oxides were calculated using the Scherrer equation from the main peak at 2θ = 33° of the XRD spectra.14,43,44 The primary particle sizes of Powder LSC1000, Pellet LSC650, Pellet LSC1000, Centrifuge LSC650, Centrifuge LSC1000 were 26.93, 12.79, 31.16, 12.35 and 28.24 nm, respectively. These results indicate that primary particle size increases with increasing calcination temperature. It has been reported that the size of synthesized solid particles increases with increasing calcination temperature due to particle aggregation.45,46 It is interesting to see that the particle size of Centrifuge LSCs is smaller than that of Pellet LSCs, regardless of calcination temperature. It is suspected that a pellet-type template prepared with a physical pressing method contains more vacant spaces than a template made with a centrifugation method.47 These spaces cause the formation of a thick wall when 3DOM LSC structures are formed, suggesting well-ordered pores in Centrifuge LSC.
FESEM images for the representative PMMA spheres and the synthesized 3DOM LSC samples are shown in Fig. 2. Fig. 2(f) confirms that the average size of PMMA spheres employed for the fabrication of the templates is about 5 ± 3 μm. For the 3DOM LSCs prepared using the pellet method at a calcination temperature of 650 °C, Pellet LSC650 (Fig. 2(b)) has thicker walls than Centrifuge LSC650 (Fig. 2(d)), but neither forms a proper 3DOM structure. Also, when calcined at 1000 °C, Pellet LSC1000 (Fig. 2(c)) shows an aggregated morphology of particles due to the disrupted skeletal structure. Fig. 2(a) shows the morphology of Powder LSC1000. Though the primary particle size of LSC1000 was 26.93 nm, the primary particles combined to create larger sized secondary particles of 200–450 nm. However, a typical 3DOM structure is observed from Centrifuge LSC samples synthesized at both 650 °C and 1000 °C. The average pore sizes of Centrifuge LSC650 and Centrifuge LSC1000 are 1.5 ± 1 μm, corresponding to 70% miniature size of PMMA (5 ± 3 μm). Thus, formed pores are interconnected three-dimensionally, and, in addition, much smaller pores are observed in each of them. This morphology of 3DOM LSCs is determined by joining of PMMA spheres arranged one-by-one in the template to form a periodic structure with regularly arranged pores. According to calcination temperature experiments, the skeleton could be formed at lower temperatures, but the size of pores for the samples calcined at lower temperatures is smaller than that for the samples calcined at higher temperatures. In the experiment range, a well-aligned 3DOM La0.6Sr0.4CoO3−δ structure without impurities was synthesized at a calcination temperature of 1000 °C.
 | ||
| Fig. 2 SEM images of 3DOM La0.6Sr0.4CoO3−δ perovskites: (a) Powder LSC1000, (b) Pellet LSC650, (c) Pellet LSC1000, (d) Centrifuge LSC650, (e) Centrifuge LSC1000 and (f) PMMA template. | ||
The specific surface areas of the samples were measured using a BET technique. 3DOM samples showed much higher specific surface area than Powder LSC1000. The surface area of Powder LSC1000 was 0.2 m3 g−1, while Pellet LSC1000 and Centrifuge LSC1000 showed much higher surface areas of 1.156 m3 g−1 and 1.4 m3 g−1, respectively, as listed in Table 1.
| Sample | Specific surface area (m2 g−1) | ORR | OER | ||
|---|---|---|---|---|---|
| Onset potential (V) | Limiting current (mA) | Onset potential (V) | Current (mA) at 1.0 V | ||
| a Ketjenblack EC600JD, the surface area data was provided from Akzo Nobel, Netherlands. | |||||
| Powder LSC1000 | 0.20 | −0.143 | 0.962 | 0.821 | 3.45 |
| Pellet LSC1000 | 1.15 | −0.135 | 1.03 | 0.790 | 4 |
| Centrifuge LSC1000 | 1.4 | −0.114 | 1.09 | 0.775 | 4.79 |
| KBa | 1410 | −0.173 | 0.759 | 0.849 | 0.32 |
3.2 Electrochemical characteristics
The electrochemical properties of La0.6Sr0.4CoO3−δ perovskite oxides with different morphologies were investigated using cyclic voltammetry (CV) and linear sweep voltammetry (LSV) techniques. The measurements were carried out after purging O2 gas in 0.1 M KOH solutions. The CV shape and oxygen reduction peak for all synthesized samples are shown to be very similar to those observed in our previous report.14 The reduction peaks were observed around −0.2 V for all synthesized samples, though the peak currents were slightly different from one other. The peak current of Centrifuge LSC1000 (0.244 mA) is higher than that of Pellet LSC1000 (0.197 mA), while the peak current of Pellet LSC1000 is higher than that of Powder LSC1000 (0.167 mA). The trend of the peak current increase is quite similar to that of the specific surface area increase of the samples. The 3DOM samples showed much higher specific surface area than Powder LSC1000, and the surface area of Centrifuge LSC1000 was the highest among the synthesized samples. This suggests that the 3DOM structure is well formed with many pores and offers better reaction performance than that of Powder LSC1000.Fig. 3 shows the catalytic activities of the synthesized LSC structures for ORR and OER by measuring the linear sweeping voltammograms in O2-saturated 0.1 M KOH solution at a scan rate of 5 mV s−1 and an electrode rotation rate of 1600 rpm. The ORR curves were measured in a voltage range of −0.8 to 0.3 V and are shown at the bottom left of Fig. 3. The onset potential and limiting current of all the LSC catalysts are superior to those of KB measured under the same experimental condition. Pellet LSC1000 and Centrifuge LSC1000 catalysts show lower onset potentials and higher limiting currents than Powder LSC1000 (listed in Table 1). Especially, Centrifuge LSC1000 catalyst exhibits the lowest onset potential (−0.114 V) among the samples. This good catalytic activity of 3DOM LSCs is due to a favorable well-ordered pore structure of 3DOM in Centrifuge LSC1000, which facilitates the fast diffusion of oxygen and electrolyte in the O2 electrode. High availability of the catalytic active sites in the 3DOM Centrifuge LSC1000 is also considered to increase the electrochemical performance of the ORR.
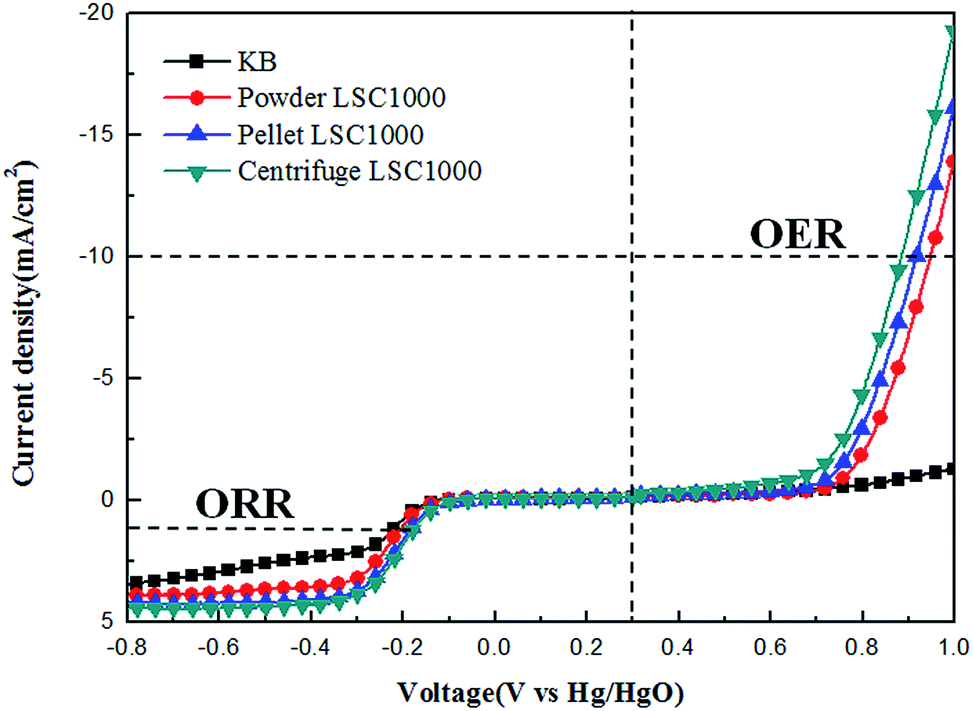 | ||
| Fig. 3 ORR and OER polarization curves of 3DOM La0.6Sr0.4CoO3−δ and KB in O2-saturated 0.1 M KOH solution at 5 mV s−1. | ||
The OER catalytic activity of the synthesized La0.6Sr0.4CoO3−δ catalysts was also studied. The polarization curves were measured during the anodic potential scan up to 1.0 V vs. Hg/HgO in 0.1 M KOH solution at a scan rate of 5 mV s−1 and a rotation rate of 1600 rpm. The OER curves, shown at the right side of Fig. 3, demonstrate that all of the synthesized perovskite catalysts exhibit much higher catalytic activity for OER than KB. This suggests that the LSC perovskites are very effective catalysts on OER.
To confirm the oxygen electrode activity, the potential difference between the ORR and OER curves was calculated from the data in Fig. 3, and the measured values are given in Table 2. The potentials of the ORR curves were selected at 1 mA cm−2, which almost approximates the half-wave potential. In addition, the potentials for the OER were chosen at the 10 mA cm−2 current density necessary to oxidize water, a convention commonly used in the OER literature.48,49 As a result, 3DOM LSC samples show lower oxygen electrode potential differences than Powder LSC1000, indicating that a 3D catalyst structure is more effective for the cathode reactions of the lithium air battery. The potential difference of Powder LSC1000 is recorded to be 1.118 V, while those of Pellet LSC1000 and Centrifuge LSC1000 are 1.093 V and 1.056 V, respectively. Centrifuge LSC1000 exhibits the lowest overpotential among all the synthesized LSC catalysts for the oxygen electrode reactions, with a value of 1.056 V. This again confirms that the 3DOM Centrifuge LSC1000 has superior catalytic activities for both ORR and OER.
| Sample | ORR: E(V) at I = 1 mA cm−2 | OER: E(V) at I = 10 mA cm−2 | Oxygen electrode Δ(OER − ORR): E(V) |
|---|---|---|---|
| Powder LSC1000 | −0.198 | 0.920 | 1.118 |
| Pellet LSC1000 | −0.176 | 0.917 | 1.093 |
| Centrifuge LSC1000 | −0.171 | 0.885 | 1.056 |
3.3 Lithium air battery application
Based on a preliminary electrochemical study of the synthesized LSC perovskite structures on ORR and OER in aqueous solution, we applied our synthesized perovskites as air cathode catalysts in a Li–air battery. The charge–discharge performance of the battery was evaluated using a Swagelok™ test cell. The cells were tested at a constant current density of 0.1 mA cm−2 in a potential range of 2.0–4.3 V at room temperature in an O2 atmosphere using 1 M LiTFSI (TEGDME) as an electrolyte. Fig. 4 presents the first discharge–charge curves measured for the synthesized perovskite catalysts, illustrating that the discharge and charge capacities of Li–O2 cells are significantly improved with the use of 3DOM LSC catalyst. Although the discharge capacity of Powder LSC1000 is recorded to be 4208 mA h g−1, those of Pellet LSC1000 and Centrifuge LSC1000 are 5316 mA h g−1 and 6066 mA h g−1, respectively. For Centrifuge LSC1000, the potential difference between the discharge and charge potential (overpotential), ΔV, measured at the discharge capacity of 1250 mA h g−1 is 0.85 V. This is a lower potential difference than those of Powder LSC1000 (1.1 V) and Pellet LSC1000 (1.09 V), which is favorable for the reversibility of the battery. This enhanced battery performance could be reasonably attributed to the synergistic effect of the catalytic activity of the 3DOM LSC catalyst. It was suspected that a favorable 3D structure of the 3DOM LSC provides a sufficient void volume for Li2O2 deposition, resulting in better reversibility of the Li–air battery. In addition, the 3DOM structure offers enough space for formation of discharge product as well as to improve transport of Li+ compared with LSC1000. | ||
| Fig. 4 Charge–discharge profiles of Powder LSC1000, Pellet LSC1000, Centrifuge LSC1000, and KB. | ||
The limited depth of discharge was examined in order to obtain complete stable cycling without any fade in the capacity. The cycle life and efficiency of the battery were measured with a limited capacity of 500 mA h g−1 at 0.1 mA cm−2, as shown in Fig. 5. The Li–O2 cells with 3DOM LSC catalyst again had better cycle stability than those with LSC1000. Especially, Centrifuge LSC1000 cycling data shows high discharge and charge cycle efficiencies up to 60 cycles and 54 cycles, respectively, without any capacity fade, unlike LSC1000 (48 and 33 cycles) and Pellet LSC1000 (53 and 51 cycles) catalysts. This enhanced cycling stability is attributed to the properties of the oxygen electrode fabricated with a well-ordered porous 3DOM LSC electrocatalyst, which facilitates the formation and decomposition of the discharge product and thus improves the rechargeability of the oxygen electrode.
 | ||
| Fig. 5 Cycle performance of Powder LSC1000, Pellet LSC1000, and Centrifuge LSC1000 catalysts under a specific capacity limit of 500 mA h g−1 at 0.1 mA cm−2: (a) discharge and (b) charge. | ||
In order to investigate the catalytic activity of the 3DOM LSC catalysts on Li2O2 decomposition, the surface composition and morphology of the air electrodes fabricated with KB, Pellet LSC1000, and Centrifuge LSC1000 catalysts were examined before and after discharge and charge processes using XRD and SEM, respectively.
Fig. S2† shows the SEM images before and after discharge/charge during the first cycle for air cathodes without and with catalysts (Pellet LSC1000 and Centrifuge LSC1000 catalysts). The SEM images before discharge show that all the air cathodes with and without catalysts show a uniform distribution with a porous surface and few noticeable differences, though the configuration of the porous surface is different. After the first discharge, regardless of catalyst, the electrode surfaces are completely covered with the agglomerated discharge solid products (see Fig. S2†). After charging, however, quite different surface morphologies are observed on the electrode surfaces with and without catalysts. The air cathode without catalysts shows a similar surface state to the discharged electrode with agglomeration, indicating poor decomposition of the solid discharge products. However, the Pellet LSC1000-catalyzed electrode exhibits nearly complete recovery of the original surface, although undecomposed solid-state discharge products are observed sporadically over the cathode surface. It is interesting that the Centrifuge LSC1000-catalyzed cathode has a surface similar to that of the original before the discharge/charge cycle, indicating almost complete decomposition of the solid discharge products. This suggests that 3DOM LSC catalysts are an effective OER catalyst for the decomposition of solid discharge products.
In order to investigate the chemical composition of the solid-state discharge products, XRD analyses were carried out for air cathodes with and without catalysts before and after the first discharge/charge cycle (Fig. S3†). XRD spectra of all cathodes show broad carbon (2θ = 23°, 44°), TAB binder (2θ = 17°), and Ni-mesh (2θ = 44.8°, 52.2.6° and 75.8°) peaks (JCPDS no. 03-1051). As observed in Fig. 1, XRD peaks related to La0.6Sr0.4CoO3 are also observed at 2θ = 23.2°, 33°, 40.7°, 47.5°, 53°, 59°, and 69.5° for the electrodes fabricated with Pellet LSC and Centrifuge LSC (JCPDS no. 36-1393).50 Before charge/discharge, the electrode without catalysts produces only the characteristic XRD peaks for KB, while the electrodes with the Pellet LSC and Centrifuge LSC catalysts have characteristic XRD peaks for La0.6Sr0.4CoO3−δ and carbon without any impurity. After the first discharge, however, the XRD spectra for KB show the formation of Li2O2 (2θ = 33°, 35° and 59°) with traces of Li2CO3 at 2θ = 21°, 31° and 32°; LiOH at 2θ = 20°, 32°, 36° and 51°; and LIOH·H2O at 2θ = 30°and 37°, respectively, as discharge products, while only Li2O2-related peaks are observed for 3DOM LSC catalysts, although the contents of the products are slightly different. However, the Li2O2-related peaks almost disappear after the recharge process. Particularly, XRD spectra for 3DOM LSC catalysts (Pellet and Centrifuge) are similar to the original spectra observed before discharge. This indicates that 3DOM LSC catalysts are effective for decomposition of the discharge product Li2O2. This coincides with the results of electrochemical characterization and battery performance studies.
In order to identify the superior catalytic activity of our 3DOM LSC catalysts for the decomposition of Li2O2 in a real Li–air battery system, CV was also measured in nonaqueous organic electrolyte solution. The measurements were carried out at a scan rate of 0.01 V s−1 over a potential range of 1.5–4.7 V at room temperature in an O2 atmosphere using 1 M LiTFSI (TEGDME) as an electrolyte. Fig. 6 shows the cyclic voltammograms of the cathodes without and with the synthesized LSC catalysts (Pellet LSC1000 and Centrifuge LSC1000). In the cathodic scan, a sharp reduction peak is observed in the ORR potential range of 3.0–2.0 V for all synthesized samples, though the peak potentials are slightly different from one other. It has been reported that O2 + Li+ + e− = LiO2 first occurs at lower potential (2.67 V) and then rapidly converts to Li2O2 through the chemical reaction of 2LiO2 = Li2O2 + O2 or the electrochemical reaction of LiO2 + Li+ + 2e− = Li2O2.51 In Fig. 4, the discharge plateau potential measured from the discharge curves of the electrodes is about 2.67 V, which corresponds to the potential measured at the FWHM of the reduction peak (2.48–2.86). This indicates that the sharp reduction peak appearing in Fig. 6 is due to the formation of Li2O2 by the ORR. For the KB electrode, the sharp peak is observed at a potential of 2.48 V, while LSC catalyst-modified electrodes show the peak at 2.53 V. This indicates that perovskite produces better ORR catalytic activity, as observed in the bottom left of Fig. 3.
 | ||
| Fig. 6 Cyclic voltammograms of the cathodes with and without the synthesized 3DOM LSC catalysts in 1 M LiTFSI (TEGDME) electrolyte at a scan rate of 0.01 V s−1 over a potential range of 1.5–4.7 V at room temperature under O2 atmosphere. | ||
In the OER potential range, sharp peaks are observed between 4.3 and 4.5 V. In order to identify the peaks, we fabricated an electrode by ball-milling commercially available powdered Li2O2 and KB. The electrode was anodically scanned in the potential rage of 2.88–4.7 V at a scan rate of 0.01 mV s−1. Anodic scan LSC shows a sharp peak around 4.44 V due to the decomposition of Li2O2, though the peak position varies slightly depending on the scan rate and electrode fabrication condition (see the inset of Fig. 6). The peak potential of the KB electrode is 4.50 V, while Pellet LSC1000 4.46 V and Centrifuge LSC1000 4.35 V. The 3DOM LSC catalyst-modified electrodes exhibit lower potentials for the decomposition of Li2O2. This clearly indicates that the 3DOM LSC catalysts are more effective than KB in the decomposition of Li2O2. This trend of catalytic activity agrees with the results observed in electrochemical studies in aqueous solution, as well as the battery performance measured in a Swagelok™-type test cell. In the cathodic scan, it has been reported that the reaction O2 + Li+ + e− = LiO2 completes through O2 + e− = O2− followed by O2− + Li+ = LiO2.51 In the anodic scan, however, the LiO2 and O2− remaining in the electrolyte are reduced to generate peaks at 3.18 V and 3.49 V, respectively, as observed by weak-intensity peaks. Due to the high reactivity of O2−, a small amount of O2− exists in the electrolyte, and a smaller peak of O2− reduction appears at 3.18 V. A similar observation was reported by Cormac O. et al.,51 who observed three peaks in the OER potential range, although the peaks appeared at different potentials compared to those of the present study because they measured the CV in different electrolytes.
4. Conclusions
The 3DOM LSC was synthesized using PMMA templates formed in two ways: arrays of pellet type using the press method and self-assembled into colloidal crystals by centrifugation. Although SrCO3 and Co3O4 impurity phases are observed in all 3DOM LSC samples prepared at calcination temperatures of 650 °C, the impurities were not observed in the spectra of the LSC synthesized at calcination temperatures of 1000 °C. The FESEM images showed that Pellet LSC650 had a thicker skeleton and was not formed with a 3DOM structure or with Centrifuge. Moreover, Pellet LSC1000 showed aggregation of particles due to the disruption of the skeleton. A perfect 3DOM perovskite La0.6Sr0.4CoO3−δ structure was formed in Centrifuge LSC samples. The electrochemical characterizations indicate that 3DOM LSC showed better catalytic performance than Powder LSC. Centrifuge LSC1000 with well-ordered 3DOM structure and no impurity exhibited the highest electrocatalytic activity and the best Li–air battery performance. The electrochemical study in nonaqueous organic electrolyte solution also demonstrated that the Centrifuge LSC1000 nanostructure was the most active catalyst among the investigated LSC structures. We concluded that the 3DOM structure contains sufficient space for the formation of discharge product over a large specific surface area and might be most active to decompose the discharge product Li2O2, leading to the reduction of overpotential by smooth transport of Li+ under charge/discharge processes. Therefore, a 3DOM perovskite La0.6Sr0.4CoO3−δ structure can be used as an oxygen electrode catalyst for the next generation of lithium–air batteries.Acknowledgements
This work was supported by the Human Resources Development program (no. 20134030200330) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government Ministry of Trade, Industry and Energy. This paper was also supported by research funds of Chonbuk National University in 2014.References
- J. Xiao, D. Wang, W. Xu, D. Wang, R. E. Williford, J. Liu and J. Zhang, J. Electrochem. Soc., 2010, 157(4), A487 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger and L. J. Hardwick, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- H. R. Bak, J. H. Lee, B. K. Kim and W. Y. Yoon, Electron. Mater. Lett., 2012, 9(2), 195 CrossRef.
- J. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- Ja-H. Ahn, J.-Y. Eom, J.-H. Kim, H. Won Kim, B. Cheol Lee and S.-S. Kim, J. Electrochem. Sci. Technol., 2015, 6(3), 75–80 CrossRef.
- S. Ho Kang, K. Song, J. Jung, M. Ru Jo, M. Alam Khan and Y.-M. Kang, J. Electrochem. Sci. Technol., 2014, 5(4), 105–108 CrossRef.
- D. Capsoni, M. Bini, S. Ferrari, E. Quartarone and P. Mustarelli, J. Power Sources, 2012, 220, 253–263 CrossRef CAS.
- J. Read, J. Electrochem. Soc., 2002, 149, A1190–A1195 CrossRef CAS.
- I. Kowalczk, J. Read and M. Salomon, Pure Appl. Chem., 2007, 79(5), 851–860 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, J. Power Sources, 2011, 196, 886–893 CrossRef CAS.
- M.-K. Song, S. Park, F. M. Alamgir, J. Cho and M. Liu, Mater. Sci. Eng., R, 2011, 72, 203–252 CrossRef.
- Z. Shao and S. M. Haile, Nature, 2004, 431, 170 CrossRef CAS.
- X. Han, Y. Hu, J. Yang, F. Cheng and J. Chen, Chem. Commun., 2014, 50, 1497 RSC.
- M. Young Oh, J. S. Jeon, J. J. Lee, P. Kim and K. S. Nahm, RSC Adv., 2015, 5, 19190–19198 RSC.
- Y. Zhao, L. Xu, L. Mai, C. Han, Q. An, X. Xu, X. Liu and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19569–19574 CrossRef CAS PubMed.
- N. Sun, H. Liu, Z. Yu, Z. Zheng and C. Shao, Solid State Ionics, 2014, 268, 125–130 CrossRef CAS.
- Z. Fu, X. Lin, T. Huang and A. Yu, J. Solid State Electrochem., 2012, 16, 1447 CrossRef CAS.
- Ji-J. Xu, D. Xu, Z.-L. Wang, H.-G. Wang, L.-L. Zhang and X.-B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3887 CrossRef CAS.
- K.-N. Jung, J.-H. Jung, W. Bin Im, S. Yoon, K.-H. Shin and J.-W. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 9902 CAS.
- H. Ohkuma, I. Uechi, N. Imanishi, A. Hirano, Y. Takeda and O. Yamamoto, J. Power Sources, 2013, 223, 319 CrossRef CAS.
- H. Minowa, M. Hayashi, M. Takahashi and T. Shodai, Electrochemistry, 2010, 78(5), 353 CrossRef CAS.
- C. Jin, Z. Yang, X. Cao, F. Lu and R. Yang, Int. J. Hydrogen Energy, 2014, 39, 2526 CrossRef CAS.
- Ji-J. Xu, Z.-L. Wang, D. Xu, F.-Z. Meng and X.-B. Zhang, Energy Environ. Sci., 2014, 7, 2213 CAS.
- Z. Du, P. Yang, L. Wang, Y. Lu, J. B. Goodenough, J. Zhang and D. Zhang, J. Power Sources, 2014, 265, 91 CrossRef CAS.
- M. Ghaffari, P. Y. Tan, M. E. Oruc, O. K. Tan, M. S. Tse and M. Shannon, Catal. Today, 2011, 161, 70–77 CrossRef CAS.
- Q. S. Song, C. H. Chiu and S. L. I. Chan, J. Appl. Electrochem., 2006, 36, 97–103 CrossRef CAS.
- H. Inoue, T. Ueda, S. Nohara, N. Fujita and C. Iwakura, Electrochim. Acta, 1998, 43(14–15), 2215–2219 CrossRef CAS.
- F. Lu, Y. Wang, C. Jin, F. Li, R. Yang and F. Chen, J. Power Sources, 2015, 293, 726–733 CrossRef CAS.
- Ji-J. Xu, Z.-L. Wang, D. Xu, F.-Z. Meng and X.-B. Zhang, Energy Environ. Sci., 2014, 7, 2213 CAS.
- H. Ullmann, N. Trofimenko, F. Tietz, D. Stover and A. Ahmad-Khanlou, Solid State Ionics, 2000, 138, 79 CrossRef CAS.
- T. Kawada and H. Yokokawa, Key Eng. Mater., 1996, 187, 125–126 Search PubMed.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397 CrossRef CAS.
- L.-W. Tai, M. M. Nasrallah, H. U. Anderson, D. M. Sparlin and S. R. Sehlin, Solid State Ionics, 1995, 76, 259 CrossRef CAS.
- O. D. Velev and E. W. Kaler, Adv. Mater., 2000, 7, 12 Search PubMed.
- D. Zou, S. Ma, R. Wan, M. Park, L. Sun, J. J. Aklonis and R. Salovey, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 137 CrossRef CAS.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 10, 12 Search PubMed.
- Z. Lei, J. Li, Y. Zhangb and S. Lu, J. Mater. Chem., 2000, 10, 2629 RSC.
- H. Yan, C. F. Blanford, B. T. Holland, M. Parent, W. H. Smyrl and A. Stein, Adv. Mater., 1999, 11, 12 CrossRef.
- S. Sakamoto and B. Dunn, J. Mater. Chem., 2002, 12, 2859 RSC.
- C. J. Patrissi and C. R. Martin, J. Electrochem. Soc., 1999, 146, 3176 CrossRef CAS.
- M. P. Pechini, US Pat., 3330697, 1967.
- K. D. Budd and D. A. Payne, Mater. Res. Soc. Symp. Proc., 1984, 32, 239 CrossRef CAS.
- P. Scherrer and N. G. W. Gottingen, Math.-Phys. Kl., 1918, 2, 96 Search PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102 CrossRef CAS.
- P. Hjalmarsson, M. Søgaard and M. Mogensen, Solid State Ionics, 2008, 179, 422 Search PubMed.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Solid State Ionics, 2012, 208, 43 CrossRef CAS.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 12, 693 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS PubMed.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397 CrossRef CAS.
- T. Nitamori, S. Kurihara and M. Misono, J. Catal., 1986, 98(1), 221 CrossRef.
- C. O. Laoire, S. Mukerjee and K. M. Abraham, J. Phys. Chem. C, 2010, 114, 9178 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02459a |
| This journal is © The Royal Society of Chemistry 2016 |