
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A base promoted multigram synthesis of aminoisoxazoles: valuable building blocks for drug discovery and peptidomimetics†
Bohdan A. Chalyka,
Inna Y. Kandaurovab,
Kateryna V. Hrebeniukb,
Olga V. Manoilenkoa,
Irene B. Kulikd,
Rustam T. Iminova,
Vladimir Kubyshkine,
Anton V. Tverdokhlebov*a,
Osman K. Ablialimov*a and
Pavel K. Mykhailiuk*ac
aEnamine Ltd., Chervonotkatska 78, 01103 Kyiv, Ukraine. Web: http://www.enamine.netE-mail: A.tverdohlebov@mail.enamine.net; O.ablialimov@mail.enamine.net; Pavel.Mykhailiuk@gmail.com; Pavel.Mykhailiuk@mail.enamine.net
bDepartment of Chemistry, Kyiv Polytechnic Institute, Prosp. Peremohy 37, Kyiv 03056, Ukraine
cDepartment of Chemistry, Taras Shevchenko National University of Kyiv, Volodymyrska Street 64, Kyiv 01601, Ukraine
dInstitute of Bioorganic Chemistry & Petrochemistry, NAS of Ukraine, Murmanska 1, Kyiv 02660, Ukraine
eInstitute of Chemistry, Technical University of Berlin, Müller-Breslau-Str. 10, Berlin 10623, Germany
First published on 1st March 2016
Abstract
A practical multigram metal free synthesis of isoxazole-containing building blocks from commercially available amino acids was elaborated. The key reaction was a regioselective [3 + 2]-cycloaddition of in situ generated nitrile oxides with alkynes/enamines. The obtained building blocks were used in the preparation of bioactive compounds and peptidomimetics.
Introduction
The isoxazole core is a structural element in many commercial pharmaceuticals (Fig. 1). The range of biological activities of the isoxazole-containing drugs is broad: analgesic,1 anti-inflammatory,1a,c,2 anthelmintic,3 anti-depressant,4 anti-bacterial,5 anticancer,6 insecticidal,7 nootropic,8 anxiolytic,8a neuroprotective,9 and HIV-inhibitory agents.10 In this context, the development of practical methods towards isoxazole-based building blocks from common cheap starting materials is truly desirable.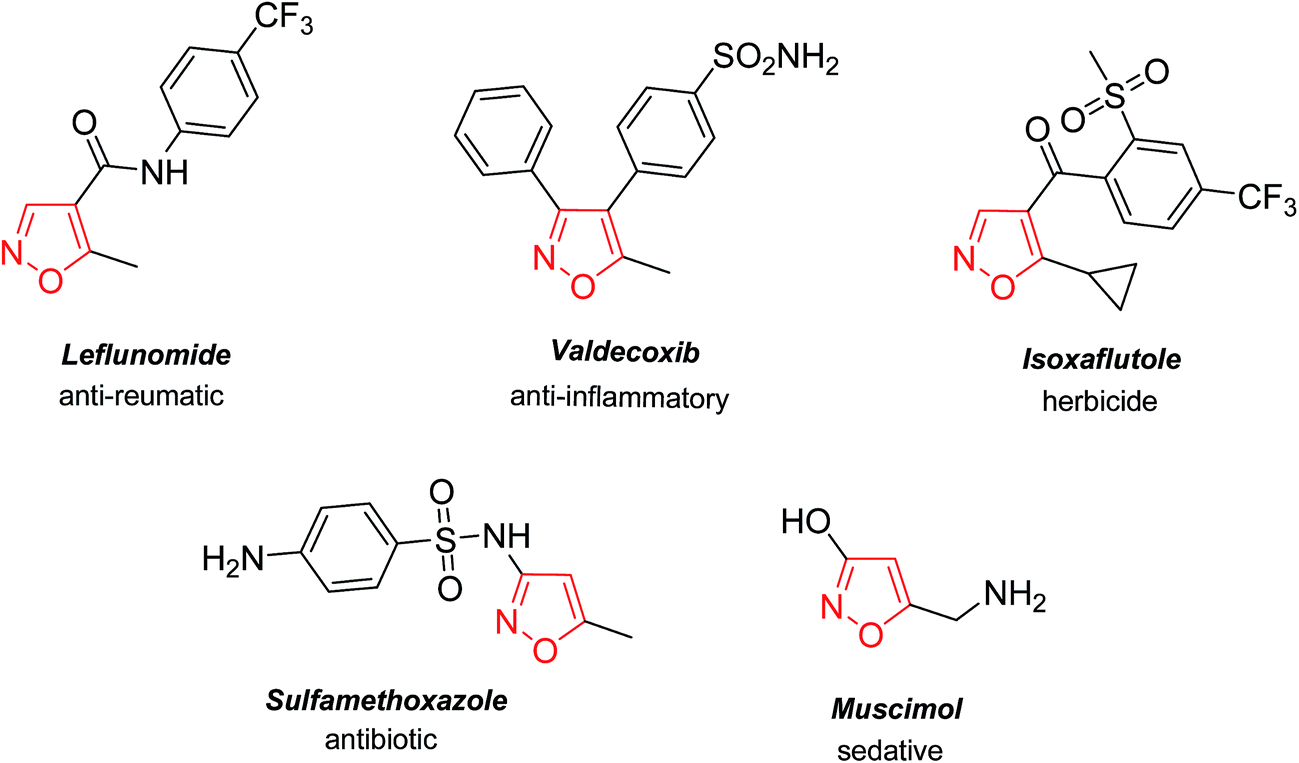 | ||
| Fig. 1 Commercialized biologically active derivatives of isoxazole. | ||
Nitrile oxides are useful intermediates towards the synthesis of a wide plethora of isoxazole-based architectures.11a–h However; most of these methods either utilize toxic metals or hypervalent iodine compounds11h which in turn, are not environmentally friendly. At the same time, there are just a few known reports dealing with metal-free conditions and most common are based on the use of triethylamine12a–b and sodium perchlorate/triethylamine.12c Therefore, the development of metal-free protocols towards the synthesis of compounds bearing isoxazole core is of particular interest.12d,e
The use of amino acids13a–h in construction of isoxazole-containing compounds has been known before. However, these reports, dealt with the specific amino acids, and the studies have not been performed systematically on large variety of substrates. Taking into account the need of exploration of metal-free protocols as well as search for easily available substrates, herein we would like to elaborate on a general practical multigram metal-free synthesis of isoxazole-core building blocks starting from commonly available and diverse α-, β- and γ-amino acids.
Results and discussion
Synthesis
|
|||||
|---|---|---|---|---|---|
| Boc-amino acid | Chloroxime | Yielda | Storageb | ||
| a Overall yield over four steps.b The products were stored in closed flasks at the room temperature.c The products were stored at 0 °C under argon. After six months ca. 10% decomposition was found according to 1H NMR. | |||||
| Boc-Gly | 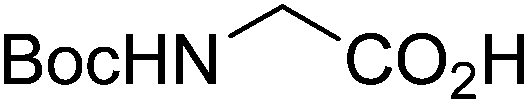 |
1 | 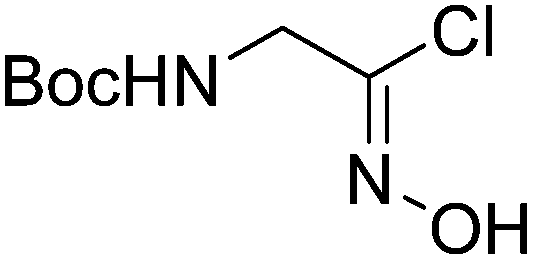 |
49% | >2 years |
| Boc-(S)-Ala | 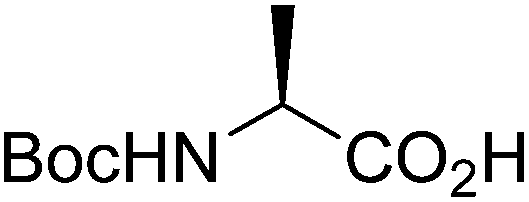 |
2 | 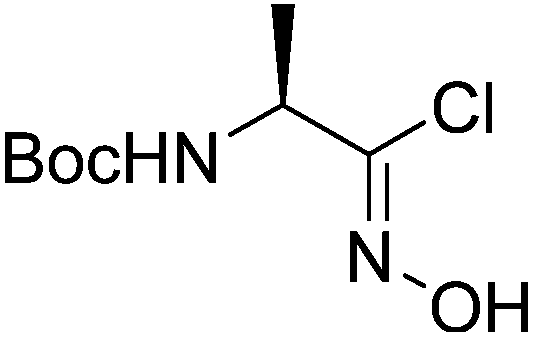 |
68% | ≤0.5 yearsc |
| Boc-(R)-Ala | 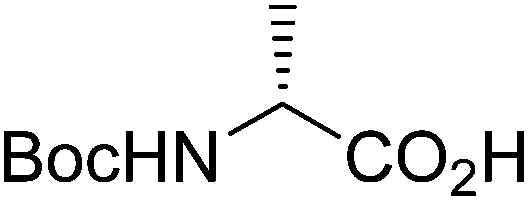 |
3 | 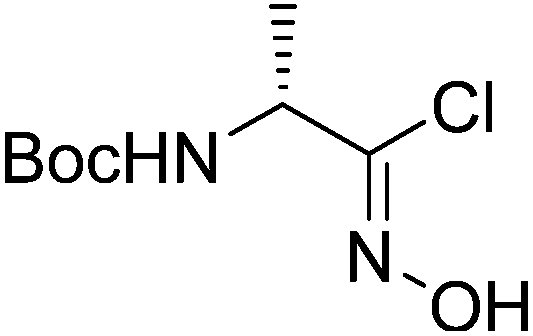 |
67% | ≤0.5 yearsc |
| Boc-(S)-Pro | 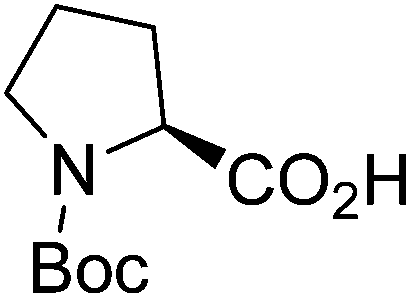 |
4 | 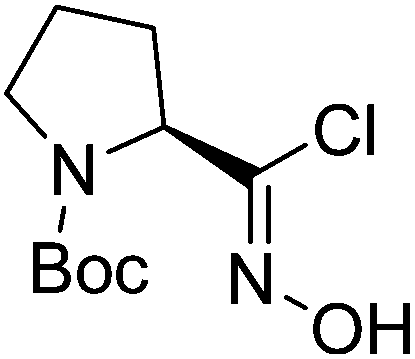 |
70% | >2 years |
| Boc-(R)-Pro | 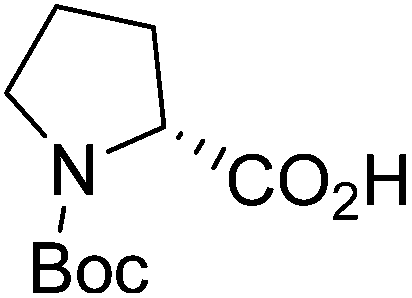 |
5 | 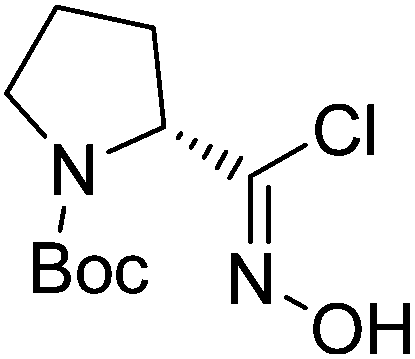 |
71% | >2 years |
| Boc-Aze | 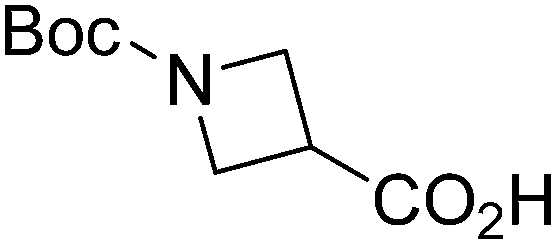 |
6 | 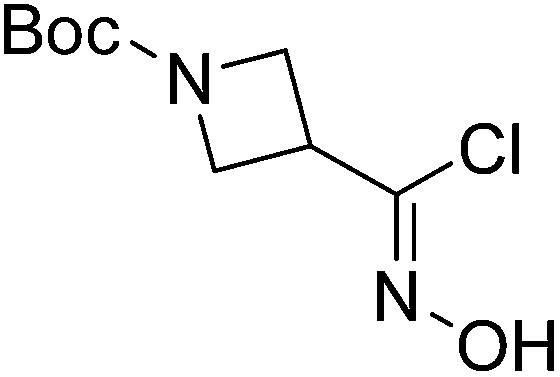 |
65% | >2 years |
| Boc-Pip |  |
7 | 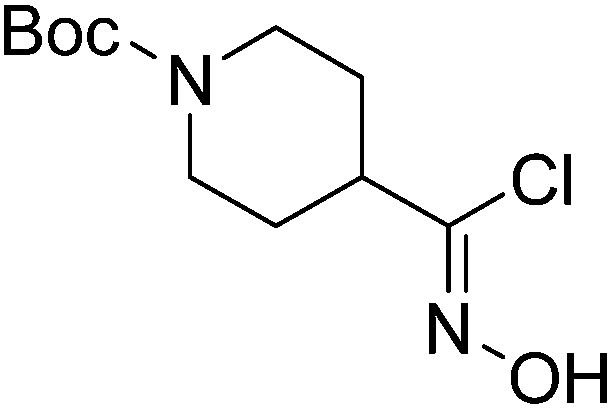 |
73% | >2 years |
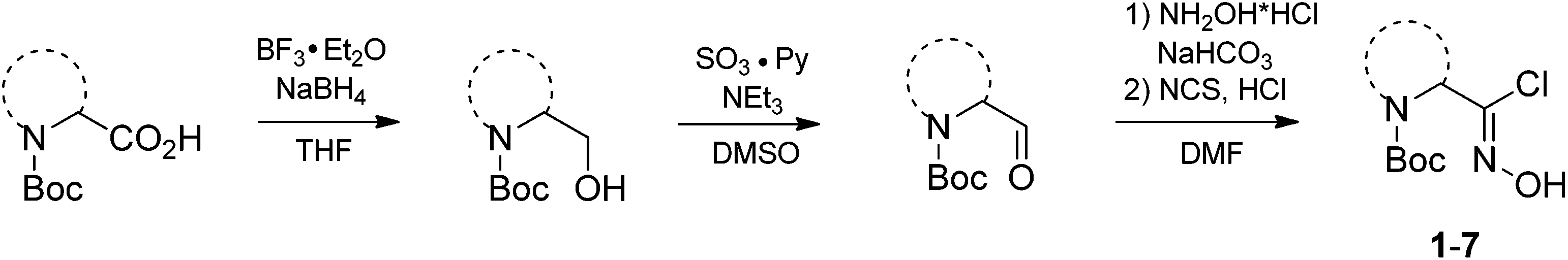
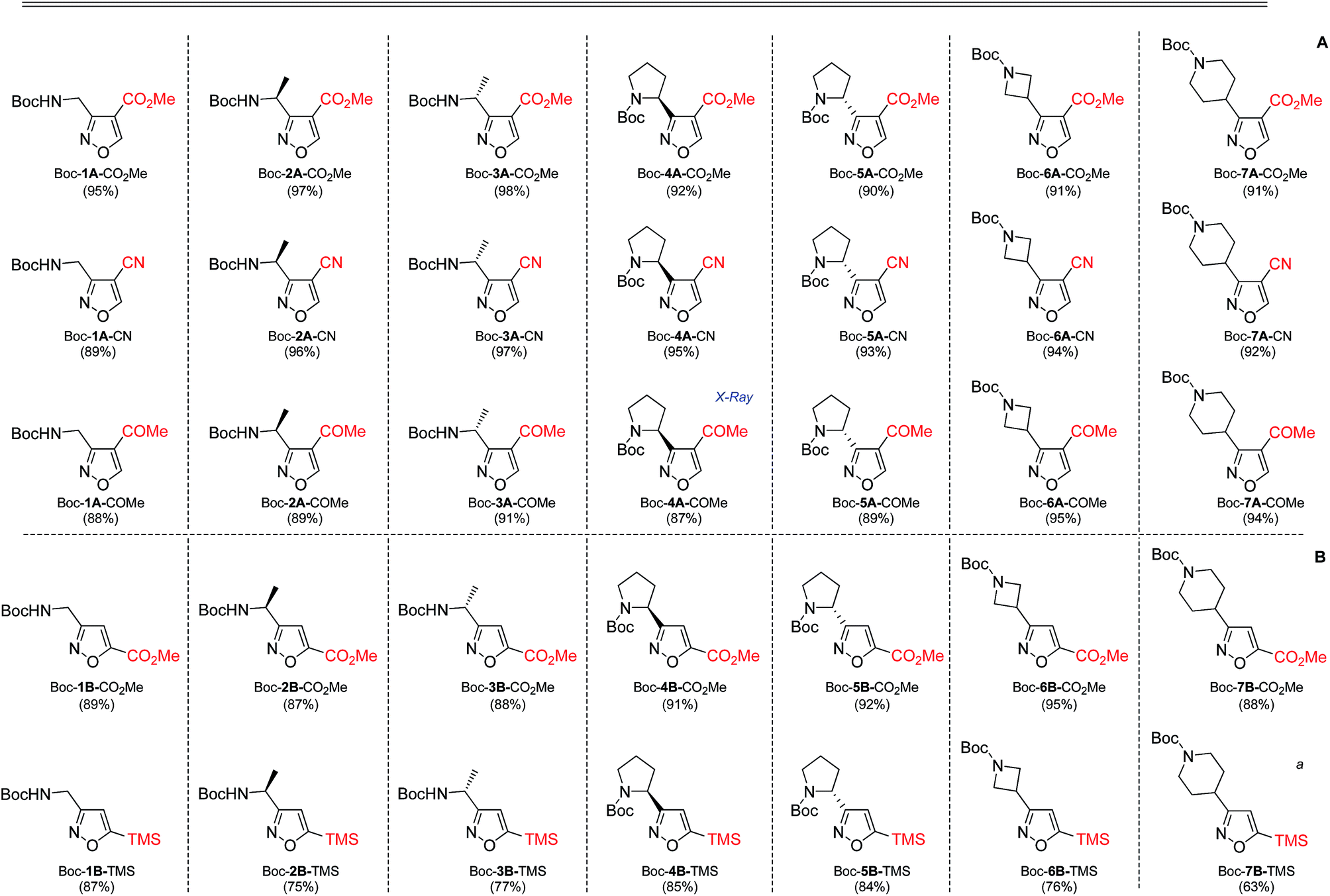
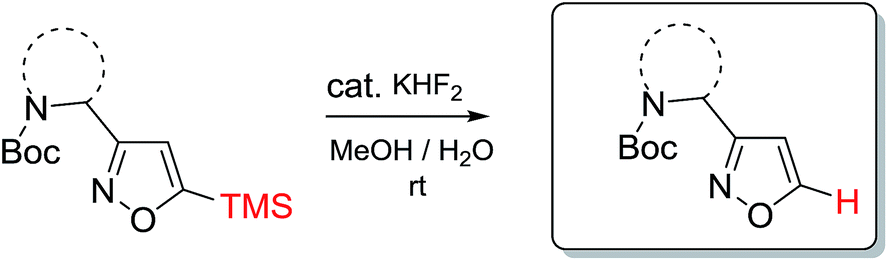
In our case, synthesis of isoxazoles from the corresponding in situ generated nitrile oxides and alkynes/enamines was analogous to the mentioned above methods. Namely, treatment of chloroximes 1–7 with a mild base (sodium hydrogen carbonate or triethylamine) at either the room temperature or at 0 °C led to generation of corresponding nitrile oxides (Table 2). Subsequent addition of enamines 8–10 (1.2 eq.) to these in situ generated nitrile oxides lead to [3 + 2]-cycloaddition intermediates that readily eliminated dimethylamine to give the 3,4-disubstituted isoxazoles in a regioselective fashion as previously reported with push–pull enamines (Table 2, A).20 In contrast, terminal alkynes 11, 12 afforded a mixture of 3,4-/3,5-isomers ∼30/70 when reaction was performed at the room temperature.21 Nevertheless, the same reaction at 0 °C exhibited an improved regioselectivity of ∼10/90, and the major 3,5-disubstituted isomers were easily isolated by column chromatography.22
|
|---|
| a Et3N was used instead of NaHCO3 to generate the nitrile oxide, 5 eq. of 12 were used. |
 |
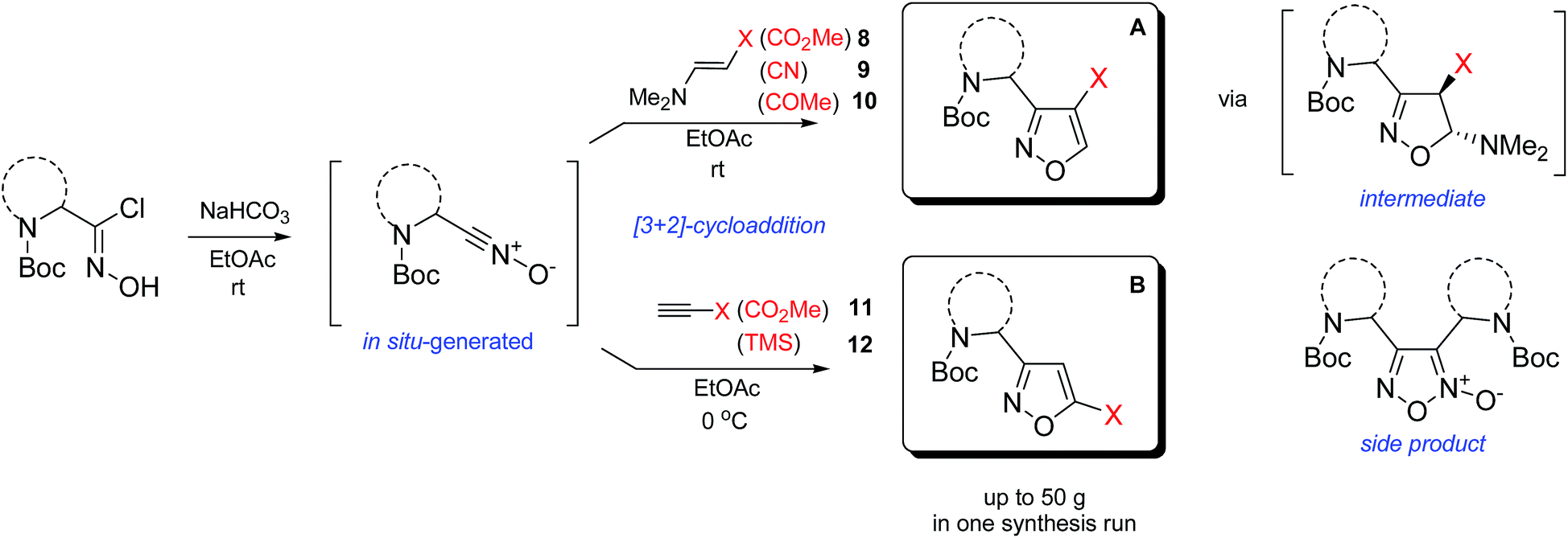
While the above described [3 + 2] cycloaddition reactions were performed in ethyl acetate the synthesis also worked well in tetrahydrofuran, dichloromethane, dimethoxymethane or chloroform, though with compromised regioselectivity. At the same time synthesis of isoxazoles in water as environmentally benign solvent is of high interest.23 It is also worth mentioning, that these reactions were performed under obligatory high dilution – substrate/solvent = 1/10 (wt/v) – otherwise formation of side furoxans was observed.24
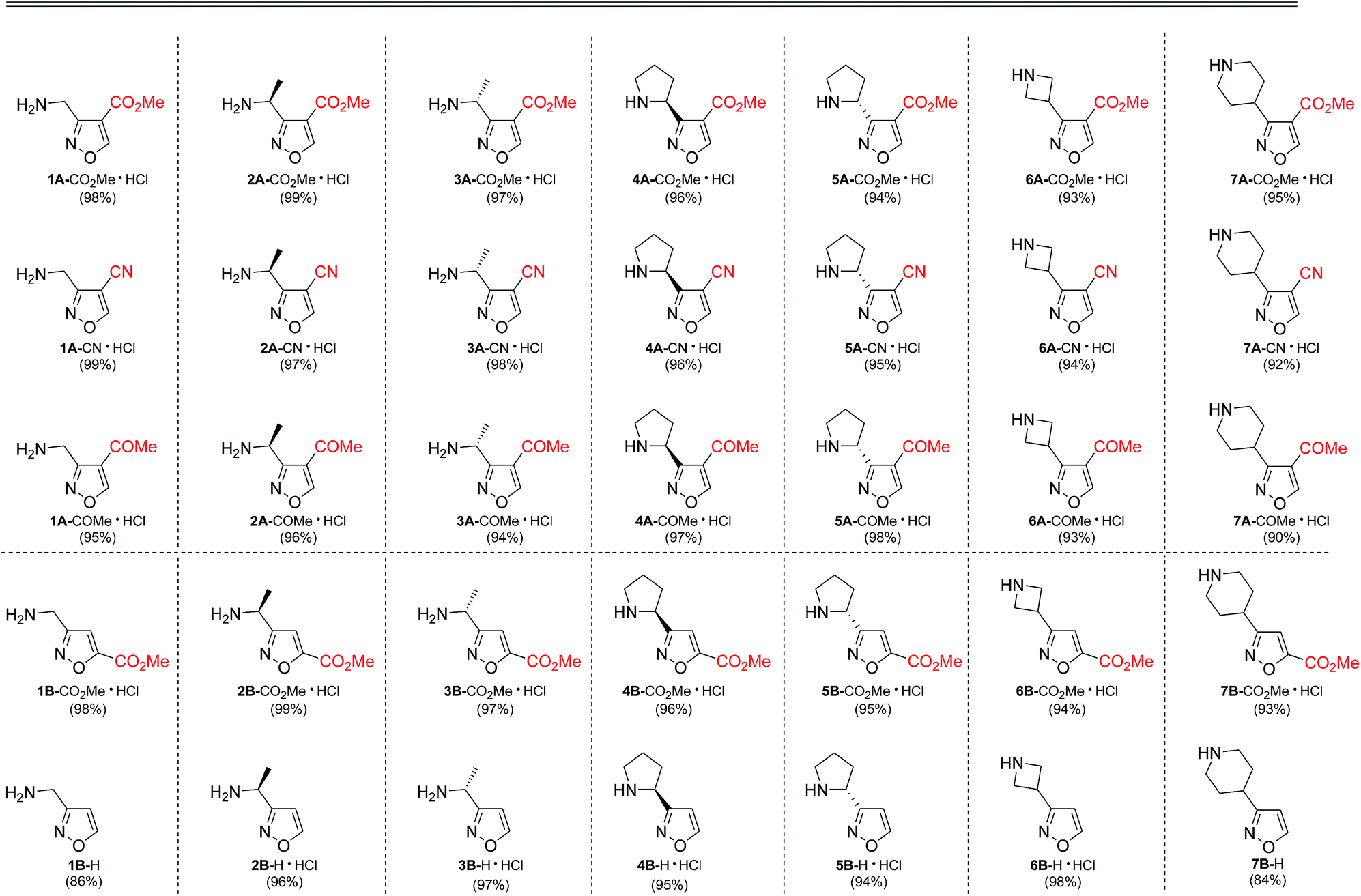
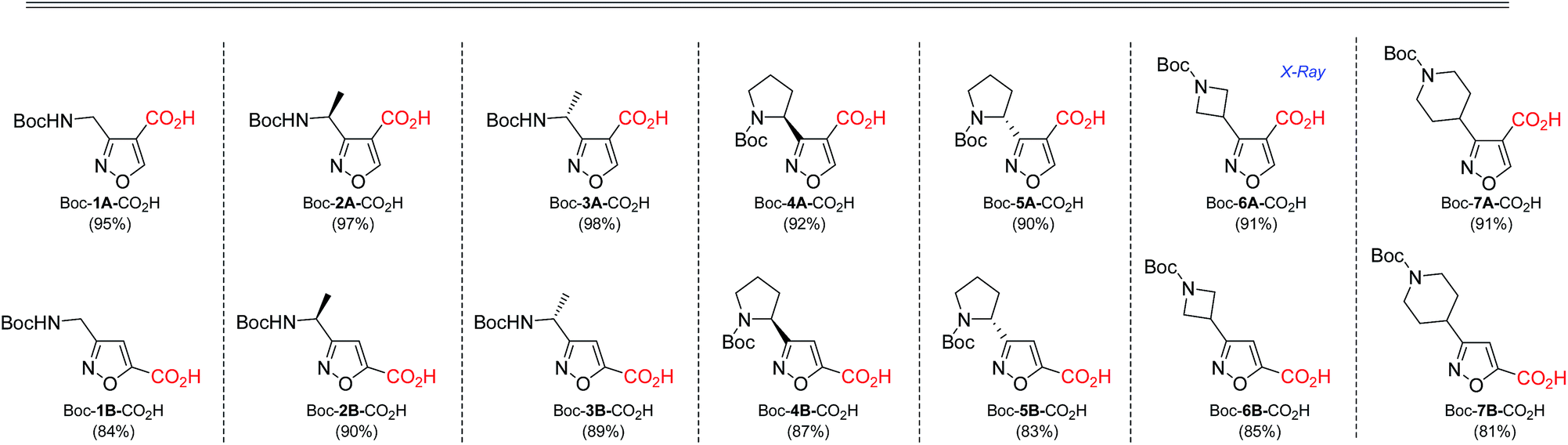
As the result we obtained a variety of N-Boc aminoisoxazoles initially starting from amino acids, and the synthetic approach was scalable such that up to 50 g of a product (Boc-6-H) was obtained in a single synthesis run.
|
|---|
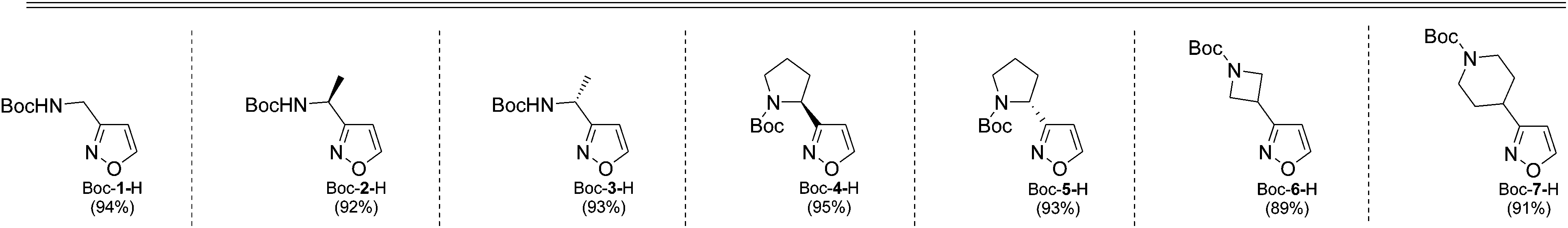 |
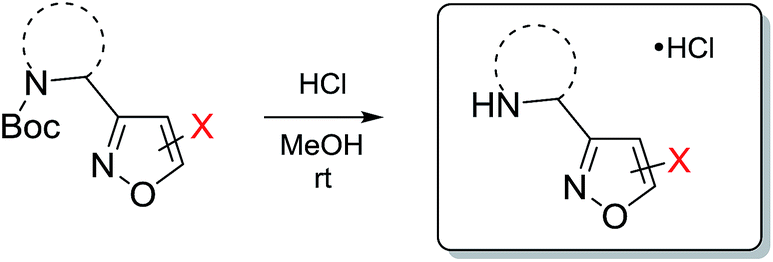
As next, cleavage of N-Boc group from all compounds was performed under acidic conditions to afford the target building blocks – aminoisoxazoles (Table 4).
|
|---|
 |
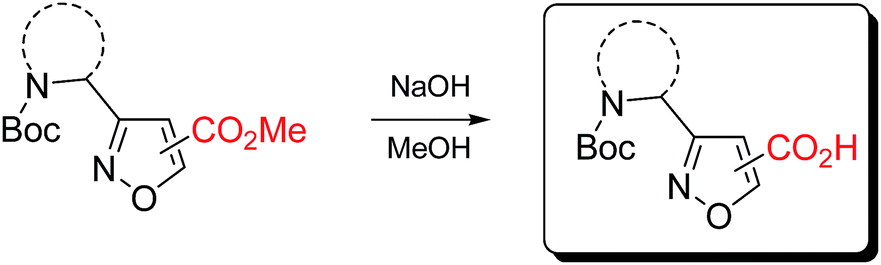
Carboxymethyl-substituted isoxazoles were saponificated with sodium hydroxide in methanol to afford corresponding free amino acids in good yields (Table 5).
|
|---|
 |
The structures of two final compounds were confirmed by X-ray analysis (Fig. 2). Interestingly, the structure of Boc-4A-COMe indicates a repulsion occurring between the carbamate carbonyl oxygen and the isoxazole nitrogen, similar to what has been recently reported for analogous structures with 1,3-oxa/thiazine in place of the isoxasole.26
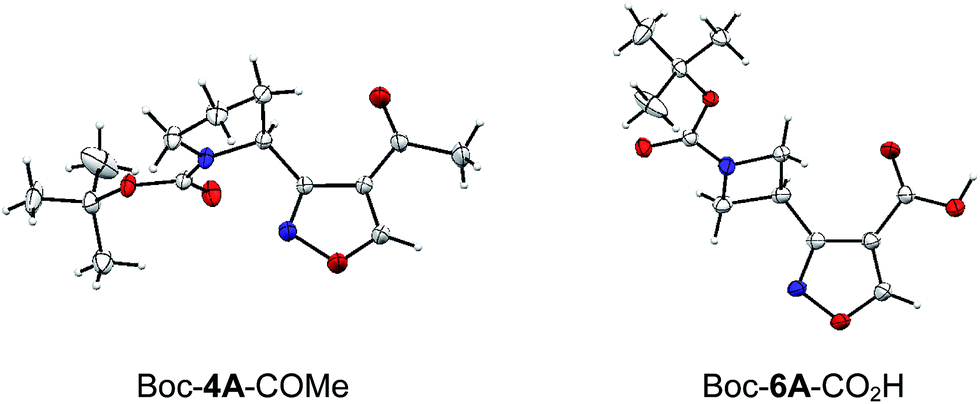 | ||
| Fig. 2 X-ray crystal structures of Boc-4A-COMe and Boc-6A-CO2H. | ||
Practical applications
After having established a practical general synthetic protocol towards isoxazole-containing building blocks, we also aimed to demonstrate applicability of the synthesized structures in other chemical areas.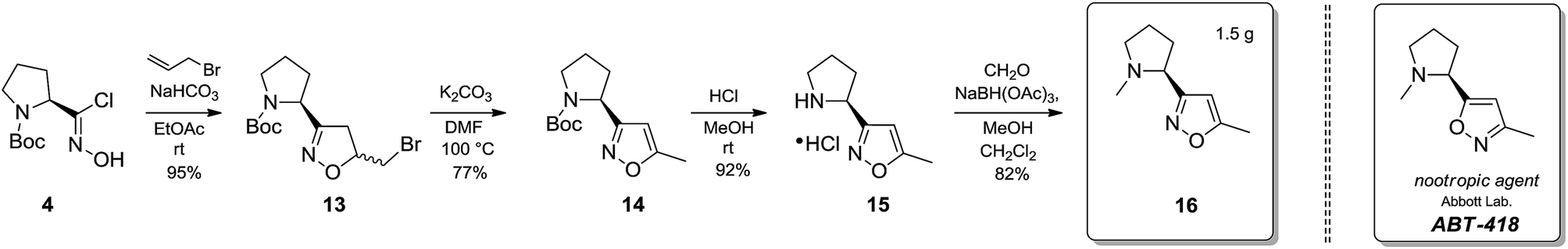 | ||
| Scheme 1 Synthesis of isoxazole 16 – bioisostere of the known nootropic agent ABT-418. | ||
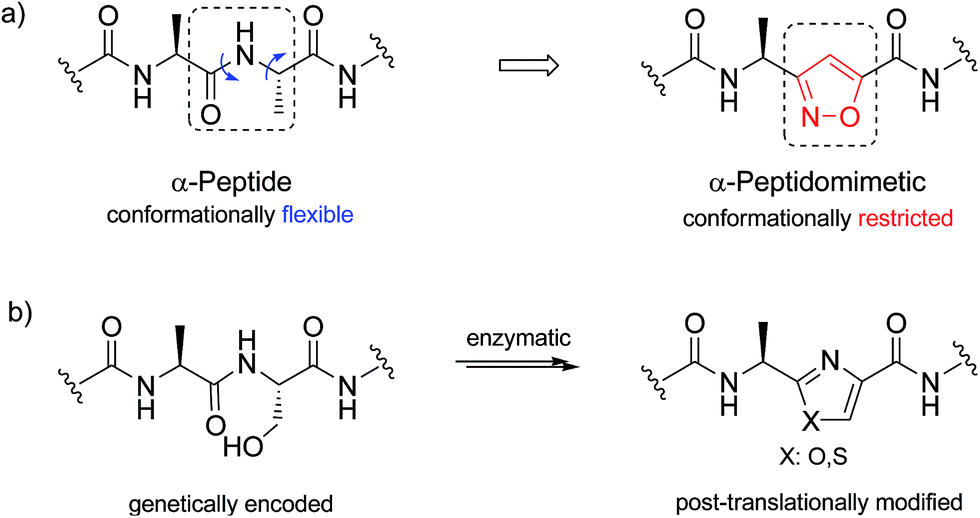 | ||
| Scheme 2 (a) α-Peptide, and its conformationally-restricted analogue with isoxazole backbone; (b) construction of a post-translationally modified peptide with oxa/thiasole backbone. | ||
In addition, isomeric oxazoles and thiazoles are common in the backbones30 of various ribosomally synthesized and post-translationally modified peptides.31 Construction of an azole ring is generally afforded in nature by cyclization of serine or threonine side chain on the downstream amide bond.32–34 Total synthesis of respective peptidomimetics has been developed in order to provide naturally occurring antimicrobial agents or toxins as well as their chemically modified analogues.35,36 Though, oxa/thiazole to isoxazole mutations have not been explored. Therefore, next we attempted the synthesis of isoxazole-containing peptidomimetics by solid phase peptide synthesis (SPPS) in order to highlight this opportunity.
Initially we synthesized the Fmoc-protected amino acid Fmoc-AlaIso-OH (17) (Scheme 3). Peptide chains were grown on Rink-amide resin, and TBTU/HOBt/DIPEA system was used for coupling. Only in the case of Ac-Leu-AlaIso-Val-NH2 (18) we observed minor degree of epimerization (≤15%), whereas in Ac-Leu-Val-AlaIso-NH2 (19) no racemization was observed.37 In addition, no degradation was found after cleavage from the resin upon treatment with TFA/TIS. Generally the amino acid Fmoc-AlaIso-OH turned out to be compatible with the standard SPPS, allowing synthesis and isolation of both target peptides by conventional methods.38
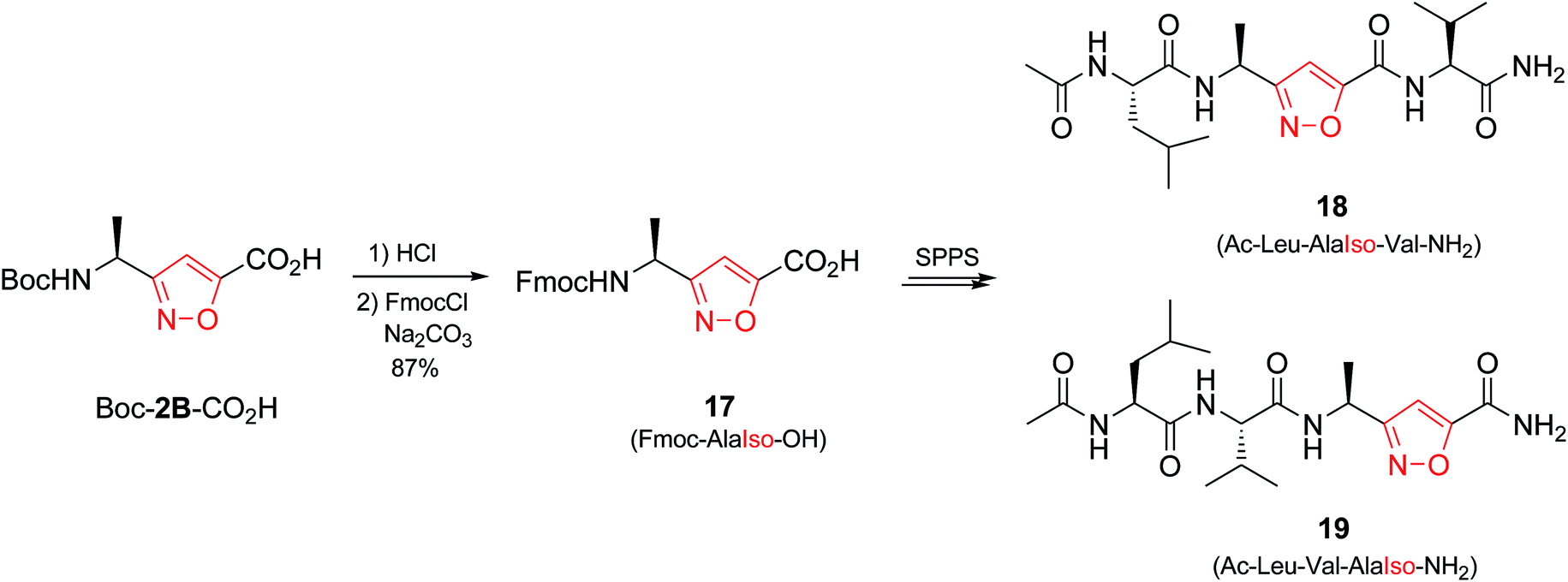 | ||
| Scheme 3 Synthesis of isoxazole-containing peptidomimetics 18 and 19. | ||
Conclusions
We have developed a general metal-free practical protocol towards isoxazole-containing building blocks starting from commonly available α-, β- and γ-amino acids. The target products were obtained in up to 50 g scale. The high potential of the developed method was demonstrated by synthesis of compound 16 – bioisostere of the known nootropic agent ABT-418, – and preparation of isoxazole-containing peptidomimetics 18, 19 by solid phase peptide synthesis.Experimental section
General
All reagents were available from Enamine Ltd. Solvents were purified according to standard procedures. When organic solutions were concentrated under reduced pressure, 35–40 °C bath temperature was used. Column chromatography was performed with silica gel 60 (230–400 mesh) as the stationary phase. 1H, 13C NMR spectra were recorded at the NMR spectrometers operating at 400 and 500 1H frequency (101 and 126 MHz for 13C experiments). NMR chemical shifts are reported in ppm, in the δ scale and are referenced using residual NMR solvent peaks at 7.26 and 77.16 ppm for 1H and 13C in CDCl3, 2.50 and 39.52 ppm for 1H and 13C in DMSO-d6, and 4.79 ppm for 1H in D2O. For peptides NMR analysis was performed in 5 mM peptide solutions in CD3OD at 700 MHz (1H frequency) spectrometer. 1H NMR spectra were recorded 25 min after the peptides were dissolved (fresh) and therefore contained few of the non-exchanged amide resonances. The 1H NMR spectra were in addition recorded after overnight measurements when the NH-to-ND exchange was already completed. 1H HOHAHA (dipsi2 of 60 ms) and ROESY (spin-lock 300 ms) spectra were recorded to complete the assignment of the 1H resonances. 13C {1H} dept45 and 1H {13C} HSQC experiments were performed in order to assign accompanied 13C resonances. The following abbreviations are used in reported NMR data: s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet), br (broad), brs (broad singlet). Coupling constants (J) are in Hz. Spectra are reported as follows: chemical shift (δ, ppm), multiplicity, integration, coupling constants (Hz). Measured melting points are uncorrected. Boiling points were measured at 0.8 mmHg unless otherwise specified herein. LC-MS data were acquired on Agilent 1200 HPLC system equipped with DAD/ELSD/LCMS-6120 diode-matrix and mass-selective detector, column: Poroshell 120 SB-C18, 4.6 mm × 30 mm. Eluent, A, acetonitrile–water with 0.1% of FA (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); B, water with 0.1% of FA. Optical rotations were measured on polarimeter in methanol using 1 dm cell; optical rotation values are given in 10−1 deg cm2 g−1; concentrations (c) are given in mmol L−1, wavelength 589 nm at 20 °C. The enantiomeric excess and retention time (tR) was determined for major signal by HPLCs: Daicel CHIRALPACK IA, 5 μm, 4.6 × 250 mm, Daicel CHIRALPACK IB, 5 μm, 4.6 × 250 mm, Daicel CHIRALPACK OJ-H, 5 μm, 4.6 × 250 mm, Daicel CHIRALPACK AS-H, 5 μm, 4.6 × 250 mm chiral columns, injection volume 0.1 μL, eluent (hexanes: 2-propanol). Solid compounds were recrystallized from acetonitrile unless other is specified.
:1); B, water with 0.1% of FA. Optical rotations were measured on polarimeter in methanol using 1 dm cell; optical rotation values are given in 10−1 deg cm2 g−1; concentrations (c) are given in mmol L−1, wavelength 589 nm at 20 °C. The enantiomeric excess and retention time (tR) was determined for major signal by HPLCs: Daicel CHIRALPACK IA, 5 μm, 4.6 × 250 mm, Daicel CHIRALPACK IB, 5 μm, 4.6 × 250 mm, Daicel CHIRALPACK OJ-H, 5 μm, 4.6 × 250 mm, Daicel CHIRALPACK AS-H, 5 μm, 4.6 × 250 mm chiral columns, injection volume 0.1 μL, eluent (hexanes: 2-propanol). Solid compounds were recrystallized from acetonitrile unless other is specified.
Synthesis of 3,4-disubstituted isoxazoles, representative procedure
1H NMR (500 MHz, DMSO-d6): δ 9.59 (s, 1H, isoxaz), 7.24 (brs, 1H, NH), 4.41 (d, 2H, CH2), 3.80 (s, 3H, OCH3), 1.39 (s, 9H, CH3-Boc). 13C NMR (125 MHz, DMSO-d6): δ 164.9, 161.1, 160.0, 155.4, 111.9, 78.2, 51.9, 35.9, 28.1. MS (APCI) m/z [M − Boc + 1H]+ calculated for C10H13N2O5: 255.1; found: 255.4. Anal. calcd for C11H16N2O5: C, 51.56; H, 6.29; N, 10.93. Found: C, 51.35; H, 6.07; N, 10.52.
Synthesis of 3,5-disubstituted isoxazoles, representative procedure
Representative procedure for cleavage of tert-butyloxycarbonyl group
1A-COMe·HCl: 95%, white powder, mp 94–96 °C. 1H NMR (500 MHz, D2O-d2): δ 9.53 (s, 1H, isoxaz), 4.54 (s, 2H, CH2), 2.60 (s, 3H, CH3-Ac). 13C NMR (125 MHz, DMSO-d6): δ 192.0, 166.1, 155.9, 119.8, 34.4, 29.0. MS (APCI) m/z [M + 1H]+ calculated for C6H9N2O2: 141.1; found: 141.2. Anal. calcd for C6H9ClN2O2: C, 40.81; H, 5.14; N, 15.86. Found: C, 40.25; H, 5.33; N, 15.96.
Representative procedure for the hydrolysis of isoxazole-based esters
Representative procedure for the for the synthesis of 3-substituted isoxazoles
Synthesis of the ABT-418 bioisostere 16
13-S-Isomer: 95%, white powder, [α]20D = −80.00 (c = 35.42). 99.6% ee, tR = 12.2 min;
13-R-Isomer: 97%, white powder, [α]20D = +81.84 (c = 35.42), 99.8% ee, tR = 10.5 min.
1H NMR (500 MHz, CDCl3): δ 4.85–4.78 (m, 1H, isoxazoline–CH), 4.63 (brs, 1H, –pyrrolidine–CH), 3.48–3.32 (m, 4H, CH2–pyrrolidine + CH2–Br), 2.94–2.90 (m, 2H, CH2–isoxazoline), 2.17–1.91 (m, 4H, CH2–pyrrolidine), 1.46 (s, 9H, CH3-Boc). 13C NMR (125 MHz, DMSO-d6, mixture of both rotamers): δ 160.3, 159.7, 153.5, 153.1, 78.8, 78.3, 54.0, 46.3, 46.0, 38.7, 35.7, 35.2, 34.8, 30.4, 30.2, 29.6, 29.3, 28.0, 23.9, 23.0. MS (APCI) m/z [M − Boc]+ calculated for C8H13BrN2O: 235.0; found: 235.1. Anal. calcd for C13H21BrN2O3: C, 46.86; H, 6.35; N, 8.41. Found: C, 46.77; H, 6.44; N, 8.57.
14-S-Isomer: 77%, [α]20D = −78.8 (c = 3.96), 99.9% ee, tR = 9.5 min;
14-R-Isomer: 75%, [α]20D = +79.63 (c = 3.96), 99.9% ee, tR = 7.6 min.
1H NMR (500 MHz, DMSO-d6): δ 6.10 (brs, 1H, isoxaz), 4.83–4.77 (m, 1H, CH–pyrrolidine), 3.43–3.38 (m, 2H, CH2–pyrrolidine), 2.36 (s, 3H, CH3), 2.21 (br, 1H, CH–pyrrolidine), 1.85 (br, 3H, CH–pyrrolidine), 1.39, (s, 9H, CH3-Boc, major rotamer), 1.24 (s, 9H, CH3-Boc, minor rotamer). 13C NMR (125 MHz, DMSO-d6): δ 169.02, 166.4, 166.0, 153.5, 153.2, 100.4, 99.8, 78.6, 78.5, 53.3, 53.1, 46.3, 46.0, 32.6, 31.3, 28.1, 27.9, 23.6, 22.9, 11.7.
MS (APCI) m/z [M + 3H]+ calculated for C13H23N2O3: 255.3; found: 255.2. Anal. calcd for C13H20N2O3: C, 61.88; H, 7.99; N, 11.10. Found: C, 61.95; H, 7.83; N, 11.14.
15-S-Isomer, white powder, 92%, [α]20D = −21.63 (c = 53.00);
15-R-Isomer, white powder, 93%, [α]20D = +22.95 (c = 53.00).
1H NMR (500 MHz, DMSO-d6): δ 10.63 (br, 1H, NH·HCl), 9.57 (br, 1H, NH·HCl), 6.55 (s, 1H, isoxaz), 4.66 (brs, 1H, CH), 3.27–3.22 (m, 2H, CH2–pyrrolidine), 2.43 (s, 3H, CH3), 2.35–2.33 (m, 1H, CH–pyrrolidine), 2.02 (br, 3H, CH2–pyrrolidine). 13C NMR (125 MHz, DMSO-d6): δ 170.6, 160.1, 101.4, 54.0, 44.4, 29.4, 22.8, 11.9. MS (APCI) m/z [M − Cl + H]+ calculated for C8H19N2O: 153.1; found: 153.2. Anal. calcd for C8H13ClN2O: C, 50.93; H, 6.95; N, 14.85. Found: C, 50.87; H, 7.08; N, 14.89.
16-S-Isomer, 82%, [α]20D = −8.00 (c = 60.16);
16-R-Isomer, 83%, [α]20D = +7.86 (c = 60.16).
1H NMR (500 MHz, DMSO-d6): δ 6.14 (brs, 1H, isoxaz), 3.24–3.19 (t, 1H, J = 10.0, CH–pyrrolidine), 3.07–3.03 (m, 1H, CH–pyrrolidine), 2.36 (s, 3H, CH3–NH), 2.26–2.19 (m, 1H, CH–pyrrolidine), 2.11 (s, 4H, CH3 + CH), 1.84–1.71 (m, 3H, CH–pyrrolidine). 13C NMR (125 MHz, DMSO-d6): δ 169.1, 166.1, 99.7, 61.3, 56.0, 31.7, 22.4, 11.8. MS (APCI) m/z [M + H]+ calculated for C9H15N2O: 167.1; found: 167.2. Anal. calcd for C9H14N2O: C, 65.03; H, 8.49; N, 16.85. Found: C, 65.14; H, 8.33; N, 16.94.
Notes and references
- (a) S. Kankala, R. K. Kankala, P. Gundepaka, N. Thota, S. Nerella, M. R. Gangula, H. Guguloth, M. Kagga, R. Vadde and C. S. Vasam, Bioorg. Med. Chem. Lett., 2013, 23, 1306–1309 CrossRef CAS PubMed; (b) B. Frølund, A. T. Jørgensen, L. Tagmose, T. B. Stensbøl, H. T. Vestergaard, C. Engblom, U. Kristiansen, C. Sanchez, P. Krogsgaard-Larsen and T. Liljefors, J. Med. Chem., 2002, 45, 2454–2468 CrossRef; (c) S. Dadiboyena and A. Nefzi, Eur. J. Med. Chem., 2010, 45, 4697–4707 CrossRef CAS PubMed.
- J. J. Talley, D. L. Brown, J. S. Carter, M. J. Graneto, C. M. Koboldt, J. L. Masferrer, W. E. Perkins, R. S. Rogers, A. F. Shaffer, Y. Y. Zhang, B. S. Zweifel and K. Seibert, J. Med. Chem., 2000, 43, 775–777 CrossRef CAS.
- J. B. Carr, H. G. Durham and D. K. Hass, J. Med. Chem., 1977, 20, 934–939 CrossRef CAS PubMed.
- (a) J. I. Andrés, J. Alcázar, J. M. Alonso, R. M. Alvarez, M. H. Bakker, I. Biesmans, J. M. Cid, A. I. De Lucas, J. Fernández, L. M. Font, K. A. Hens, L. Iturrino, I. Lenaerts, S. Martínez, A. A. Megens, J. Pastor, P. C. M. Vermote and T. Steckler, J. Med. Chem., 2005, 48, 2054–2071 CrossRef PubMed; (b) H. S. Youn, E. J. Lee, J. E. Lee, W.-K. Park, D.-J. Baek, Y. S. Cho, H. Y. Koh, H. Choo and A. N. Pae, Bull. Korean Chem. Soc., 2009, 30, 1873–1876 CrossRef CAS.
- (a) M. Ma, Y. Cheng, Z. Xu, P. Xu, H. Qu, Y. Fang, T. Xu and L. Wen, Eur. J. Med. Chem., 2007, 42, 93–98 CrossRef CAS PubMed; (b) Ö. Temiz-Arpacı, İ. Yıldız, S. Özkan, F. Kaynak, E. Akı-Şener and İ. Yalçın, Eur. J. Med. Chem., 2008, 43, 1423–1431 CrossRef PubMed.
- (a) W.-T. Li, D.-R. Hwang, C.-P. Chen, C.-W. Shen, C.-L. Huang, T.-W. Chen, C.-H. Lin, Y.-L. Chang, Y.-Y. Chang, Y.-K. Lo, H.-Y. Tseng, C.-C. Lin, J.-S. Song, H.-C. Chen, S.-J. Chen, S.-H. Wu and C.-T. Chen, J. Med. Chem., 2003, 46, 1706–1715 CrossRef CAS PubMed; (b) J. Yong, C. Lu and X. Wu, Med. Chem. Commun., 2014, 5, 968–972 RSC.
- N. Jacobsen, L.-E. K. Pedersen and A. Wengel, Pestic. Sci., 1990, 29, 95–100 CrossRef CAS.
- (a) D. S. Garvey, J. T. Wasicak, M. W. Decker, J. D. Brioni, M. J. Buckley, J. P. Sullivan, G. M. Carrera, M. W. Holladay, S. P. Arneric and M. Williams, J. Med. Chem., 1994, 37, 1055–1059 CrossRef CAS; (b) A. Carenzi, D. Chiarino, D. D. Bella, M. Napoletano, A. Sala, US Pat. 4985428, Jan 15, 1991.
- M. Koufaki, A. Tsatsaroni, X. Alexi, H. Guerrand, S. Zerva and M. N. Alexis, Bioorg. Med. Chem., 2011, 19, 4841–4850 CrossRef CAS PubMed.
- (a) S. Srivastava, L. K. Bajpai, S. Batra, A. P. Bhaduri, J. P. Maikhuri, G. Gupta and J. D. Dhar, Bioorg. Med. Chem., 1999, 7, 2607–2613 CrossRef CAS PubMed; (b) K. A. Kumar and P. Jayaroopa, Int. J. Pharm., Chem. Biol. Sci., 2013, 3(2), 294–304 Search PubMed.
- (a) F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS; (b) M. S. Mohamed Ahmed, K. Kobayashi and A. Mori, Org. Lett., 2005, 7, 4487–4489 CrossRef PubMed; (c) T. V. Hansen, P. Wu and V. V. Fokin, J. Org. Chem., 2005, 70, 7761–7764 CrossRef CAS PubMed; (d) S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285–8287 CrossRef PubMed; (e) A. V. Dubrovskiy, P. Jain, F. Shi, G. H. Lushington, C. Santini, P. Porubsky and R. C. Larock, ACS Comb. Sci., 2013, 15, 193–201 CrossRef CAS PubMed; (f) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef; (g) T. M. Vishwanatha and V. V. Sureshbabu, J. Heterocycl. Chem., 2015, 52, 1823–1833 CrossRef CAS; (h) A. Singhal, S. K. R. Parumala, A. Sharma and R. K. Peddinti, Tetrahedron Lett., 2016, 57, 719–722 CrossRef CAS.
- (a) M. A. Weidner-Wells, T. C. Henninger, S. A. Fraga-Spano, C. M. Boggs, M. Matheis, D. M. Ritchie, D. C. Argentieri, M. P. Wachter and D. J. Hlasta, Bioorg. Med. Chem. Lett., 2004, 14, 4307–4311 CrossRef CAS PubMed; (b) S. Dadiboyena and A. Nefzi, Tetrahedron Lett., 2012, 53, 2096–2099 CrossRef CAS PubMed; (c) D. E. Kizer, R. B. Miller and M. J. Kurth, Tetrahedron Lett., 1999, 40, 3535–3538 CrossRef CAS; (d) S. Mohammed, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2015, 5, 3470–3473 RSC; (e) P. K. Mykhailiuk, Org. Biomol. Chem., 2015, 13, 3438–3445 RSC.
- (a) R. C. F. Jones, S. J. Hollis and J. N. Iley, Tetrahedron: Asymmetry, 2000, 11, 3273–3276 CrossRef CAS; (b) Y. K. Kang, K. J. Shin, K. H. Yoo, K. J. Seo, C. Y. Hong, C.-S. Lee, S. Y. Park, D. J. Kim and S. W. Park, Bioorg. Med. Chem. Lett., 2000, 10, 95–99 CrossRef CAS PubMed; (c) R. C. F. Jones, L. E. Seager and M. R. J. Elsegood, Synlett, 2011, 211–214 CrossRef CAS; (d) R. C. F. Jones and T. A. Pillainayagam, Synlett, 2004, 15, 2815–2817 CrossRef; (e) R. C. F. Jones, J. P. Bullous, C. C. M. Law and M. R. J. Elsegood, Chem. Commun., 2014, 50, 1588–1590 RSC; (f) M. Falorni, G. Giacomelli and E. Spanu, Tetrahedron Lett., 1998, 39, 9241–9244 CrossRef CAS; (g) M. Falorni, G. Giacomelli and A. M. Spanedda, Tetrahedron: Asymmetry, 1998, 9, 3039–3046 CrossRef CAS; (h) L. De Luca, G. Giacomelli and A. Riu, J. Org. Chem., 2001, 66, 6823–6825 CrossRef CAS PubMed.
- D. C. Schmitt, L. Lam and J. S. Johnson, Org. Lett., 2011, 13, 5136–5139 CrossRef CAS PubMed.
- (a) Q.-f. Jia, P. M. S. Benjamin, J. Huang, Z. Du, X. Zheng, K. Zhang, A. H. Conney and J. Wang, Synlett, 2013, 79–84 CAS; (b) S. Zhu, S. Shi and S. W. Gerritz, Tetrahedron Lett., 2011, 52, 4001–4004 CrossRef CAS.
- C. C. Lee, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2009, 7, 4349–4351 CAS.
- H. Kawai, Y. Sugita, E. Tokunaga and N. Shibata, Eur. J. Org. Chem., 2012, 1295–1298 CrossRef CAS.
- (a) S. Dadiboyena, J. Xu and A. T. Hamme II, Tetrahedron Lett., 2007, 48, 1295–1298 CrossRef CAS PubMed; (b) J. Xu and A. T. Hamme II, Synlett, 2008, 919–923 CAS.
- M. A. Schmidt, K. Katipally, A. Ramirez, O. Soltani, X. Hou, H. Zhang, B.-C. Chen, X. Qian and R. P. Deshpande, Tetrahedron Lett., 2012, 53, 3994–3997 CrossRef CAS.
- (a) A. O. Abdelhamid, A. A. Fahmi and K. N. M. Halim, Synth. Commun., 2013, 43, 1101–1126 CrossRef CAS; (b) M. R. Shaaban, T. M. A. Eldebss, A. F. Darweesh and A. M. Farag, J. Heterocycl. Chem., 2008, 45, 1739–1744 CrossRef CAS.
- (a) A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products, John Wiley & Sons, Ltd, 2002 CrossRef; (b) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef; (c) L. N. Sobenina, D. N. Tomilin, M. D. Gotsko, I. A. Ushakov, A. I. Mikhaleva and B. A. Trofimov, Tetrahedron, 2014, 70, 5168–5174 CrossRef CAS.
- (a) C. Bryant, I. D. Kerr, M. Debnath, K. K. H. Ang, J. Ratnam, R. S. Ferreira, P. Jaishankar, D. M. Zhao, M. R. Arkin, J. H. McKerrow, L. S. Brinen and A. R. Renslo, Bioorg. Med. Chem. Lett., 2009, 19(21), 6218–6221 CrossRef CAS PubMed; (b) P. Jones, M. E. Difrancesco, A. Petrocchi, C. L. Carroll, J. Marszalek, B. Czako, R. Johnson, J. Theroff, US Pat. US2015252058 (A1), Feb 27, 2014.
- L. Han, B. Zhang, M. Zhu and J. Yan, Tetrahedron Lett., 2014, 55, 2308–2311 CrossRef CAS.
- (a) T. Pasinszki, B. Hajgató, B. Havasi and N. P. C. Westwood, Phys. Chem. Chem. Phys., 2009, 11, 5263–5272 RSC; (b) G. Romeo and U. Chiacchio, in Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. J. Vaquero and J. Barluenga, Wiley-VCH Verlag & Co., Weinheim, Germany, 2011, pp. 1047–1252 Search PubMed.
- B. Guillaume, K. M. Fiona, B. Renaud, L. Clemens, Q. Laura, T. Stephan, US Pat. US2014045890 (A1), Feb 13, 2014.
- D. N. Reddy, R. Thirupathi, S. Tumminakatti and E. N. Prabhakaran, Tetrahedron Lett., 2012, 53, 4413–4417 CrossRef CAS.
- (a) S. P. Arneric, J. P. Sullivan, C. A. Briggs, D. Donnelly-Roberts, D. J. Anderson, J. L. Raszkiewicz, M. Hughes, E. D. Cadman, P. Adams, D. S. Garvey, J. Wasicak and M. Williams, J. Pharmacol. Exp. Ther., 1994, 270, 310–318 CAS; (b) D. S. Garvey, J. T. Wasicak, R. L. Elliott, S. Lebold, A.-M . Hettinger, G. M. Carrera, N.-H. Lin, Y. He, M. W. Holladay, D. J. Anderson, E. D. Cadman, J. L. Raszkiewicz, J. P. Sullivan and S. P. Arneric, J. Med. Chem., 1994, 37, 4455–4463 CrossRef CAS PubMed.
- (a) A. Plant, F. Stieber, J. Scherkenbeck, P. Lösel and H. Dyker, Org. Lett., 2001, 3, 3427–3430 CrossRef CAS PubMed; (b) I. V. Komarov, A. O. Grigorenko, A. V. Turov and V. P. Khilya, Russ. Chem. Rev., 2004, 73, 785–810 CrossRef CAS; (c) S. Pellegrino, A. Contini, M. L. Gelmi, L. Lo Presti, R. Soave and E. Erba, J. Org. Chem., 2014, 79, 3094–3102 CrossRef CAS PubMed.
- In particular, it was been recently demonstrated that terminal 2-oxasole-group is prone to induce β-conformation of the residue: (a) D. Siodłak, M. Staś, M. A. Broda, M. Bujak and T. Lis, J. Phys. Chem. B, 2014, 118, 2340–2350 CrossRef PubMed. 4,5-Thiasole-based building blocks with (R)-chirality have been used to promote β-turn structures in gramicidin (S)-analogues to construct peptidomimetics with reduced hemolytic activity: (b) B. Legrand, L. Mathieu, A. Lebrun, S. Andriamanarivo, V. Lisowski, N. Masurier, S. Zirah, Y. K. Kang, J. Martinez and L. T. Maillard, Chem.–Eur. J., 2014, 20, 6713–6720 CrossRef CAS PubMed.
- R. A. Hughes and C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 7930–7954 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- K. L. Dunbar and D. A. Mitchell, J. Am. Chem. Soc., 2013, 135, 8692–8701 CrossRef CAS PubMed.
- R. S. Roy, A. M. Gehring, J. C. Milne, P. J. Belshaw and C. T. Walsh, Nat. Prod. Rep., 1999, 16, 249–263 RSC.
- E. M. Nolan and C. T. Walsh, ChemBioChem, 2009, 10, 34–53 CrossRef CAS PubMed.
- V. S. C. Yeh, Tetrahedron, 2004, 60, 11995–12042 CrossRef CAS.
- For instance see total synthesis of plantazolicin A: (a) S. Banala, P. Ensle and R. D. Süssmuth, Angew. Chem., Int. Ed., 2013, 52, 9518–9523 CrossRef CAS PubMed ; microcin B17:; (b) R. E. Thompson, F. Collin, A. Maxwell, K. A. Joliffe and R. J. Payne, Org. Biomol. Chem., 2014, 12, 1570–1578 RSC ; sansavamide A:; (c) M. R. Davis, E. K. Singh, H. Wahyudi, L. D. Alexander, J. B. Kunicki, L. A. Nazarova, K. A. Fairweather, A. M. Giltrap, K. A. Jolliffe and S. R. McAlpine, Tetrahedron, 2012, 68, 1029–1051 CrossRef CAS PubMed ; (R)-telomestatin:; (d) T. Doi, M. Yoshida, K. Shin-ya and T. Takahashi, Org. Lett., 2006, 8, 4165–4167 CrossRef CAS PubMed ; or dendroamine A:; (e) Z. Xia and C. D. Smith, J. Org. Chem., 2001, 66, 3459–3466 CrossRef CAS PubMed.
- Curiously, epimerization of the AlaIso residue in Ac-Leu-AlaIso-Val-NH2 (but not in Ac-Leu-Val-AlaIso-NH2) would potentially lead to a β-turn structure rather than semi-extended β. This may be the driving force for the partial epimerization observed experimentally.
- G. Chennakrishnareddy, B. Vasantha, N. Narendra and V. V. Sureshbabu, Int. J. Pept. Res. Ther., 2011, 17, 185–191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization of all new compounds (PDF). CCDC [1442715 and 1442716]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra02365g |
| This journal is © The Royal Society of Chemistry 2016 |