DOI:
10.1039/C6RA02318E
(Paper)
RSC Adv., 2016,
6, 36238-36247
Application of an optimized modified stir bar with ZnS nanoparticles loaded on activated carbon for preconcentration of carbofuran and propoxur insecticides in water samples and their HPLC determination
Received
26th January 2016
, Accepted 15th March 2016
First published on 1st April 2016
Abstract
In this study, the stir bar was coated with ZnS-NPs loaded on activated carbon (AC) (ZnS-AC) as well as 1-ethyl-3-methylimidazolium hexafluorophosphate ([EMIM][PF6]) ionic liquid (IL) using a sol gel technique which was used for stir bar sorptive extraction (SBSE) of carbofuran and propoxur. The extracted analytes were then quantified by high performance liquid chromatography (HPLC) equipped with an ultra violet detector. The best extraction performance for carbofuran and propoxur was obtained through the optimization of the factors affecting SBSE including the pH of the sample solution, ionic strength, extraction time, volume of desorption solvent, desorption time, and stirring speed. The fractional factorial design (FFD) was used to find the most important factors, which were then optimized by the central composite design (CCD) and the desirability function (DF). Under the optimal experimental conditions, the proposed method has linear ranges over 0.002–30 μg mL−1 with detection limits of 0.0003–0.0005 μg mL−1 and good RSDs (and n = 6) of 3.3–4.5% with the enrichment factors (EFs) in the range of 75.6–81.6-fold for carbofuran and propoxur. The developed method has been successfully applied to the determination of two N-methylcarbamate in environmental water samples such as tap water, river water, and mineral water.
1. Introduction
N-Methylcarbamate pesticides (NMCs) were commercially available in the 1950s and since then, they have been increasingly used in modern agriculture, for example, in the production of potato, cabbage, coffee, citrus and other crops.1–3 Due to their widespread use, carbamate pesticides may enter into environmental water systems through various paths, including soil seepage, spraying, storage and the discharge of wastewater, leading to possible contamination of the environmental water.4–6 Although carbamates are generally highly biodegradable, some are hazardous to the environment and human health because carbamates are central nervous system toxins and strong endocrine disruptors, affecting humans and animals at low doses. For these reasons, carbamates and their metabolites are suspected to be carcinogens and mutagens.7,8 Therefore, it is of great importance to develop rapid and sensitive detection methods for carbamate pesticides. Generally, the target analytes in the environment are found at low concentrations within complex matrices, so sample concentration and purification prior to instrumental analysis is a key step in analytical process and the subsequent effective instrumental analysis.
However, traditional sample pretreatment such as liquid–liquid extraction (LLE) and solid phase extraction (SPE) have often confirmed to be labor-intensive, time-consuming, expensive and hazardous to health due to the large volume of potentially toxic solvents involved.
In recent years, several new environmental friendly sample pretreatment techniques, such as liquid phase microextraction (LPME),9–11 magnetic solid phase extraction (MSPE),12 solid phase microextraction (SPME)13,14 and stir bar sorptive extraction (SBSE)15,16 have been developed for the enrichment of trace analyte. Among them, SBSE has many advantages, such as high sensitivity, good reproducibility, and being organic solvent free or less, and has been successfully applied to the analysis of various analytes in environmental, food and biological samples.17,18 In SBSE (an equilibrium technique), which was introduced by Baltussen in 1999,15 the extractant is a material coated on the surface of capillary glass that plays an important role in the selectivity, polarity, adsorption capacity, and extraction efficiency. However, only polydimethylsiloxane (PDMS), ethylene glycol (EG)–silicone and polyacrylate (PA) are the commercially available coatings for SBSE presently, limiting its application in the real-world samples.17–21 One of the greatest drawbacks of commercial stir bars is the limitation in the variety of polarity of the coating materials. To cope with this limitation, some new coating materials including C16-MCM-41, molecular imprinted polymers, and polyaniline/hydroxyl multi-walled carbon nanotubes have recently been prepared and used for the extraction of polar/apolar compounds.22–25 Therefore, the development of novel stir bar coatings with high affinity and selectivity toward target analytes is of great interest. Furthermore, the glass surfaces of stir bars are in need of modification in order to immobilize the new coating on them. Conventional techniques such as monolithic materials and sol–gel technology have been applied for the modification of the coating surfaces of the stir bars.26–28 The sol–gel technology with the advantages of strong adhesion of the coating and bed, high thermal stability, high flexibility and possibility of the coating with higher surface area is the preferred one.29,30
In recent years, due to the unique properties such as excellent thermal, mechanical, and optical properties, high adsorbent capacity and specific surface area, the application of nanoparticles as the sorbents in solid phase extraction has been increased.25,26 In this work, ZnS-NPs loaded on activated carbon (AC) (ZnS-AC-NPs) were introduced as a coating materials for SBSE. The sol–gel technique was used to modify the surface of the stir bar with ZnS-AC-NPs and 1-ethyl-3-methylimidazolium hexafluorophosphate ([EMIM][PF6]) ionic liquid (IL). Then the modified stir bar with nanoparticles was applied for the extraction and preconcentration of carbofuran and propoxur from water samples prior to their determination by high performance liquid chromatography (HPLC). The influence of the important factors affecting the SBSE were investigated and optimized by the experimental design methods including of fractional factorial design (FFD), central composite design (CCD) and response surface methodology (RSM).
2. Experimental
2.1. Reagents and materials
Carbofuran and propoxur insecticide standard were purchased from Sigma-Aldrich (Steinheim, Germany). The stock solution (200 mg L−1) of carbamate insecticides were prepared by dissolving each of them in methanol and stored at 4 °C before use. The working solutions were freshly prepared through the appropriate dilution of the stock solution with double distilled/deionized water (produced by a Milli-Q system (Millipore, Bedford, MA, USA)). Tetraethyl orthosilicate (TEOS), ethanol, acetonitrile, methanol, dichloromethane, acetic acid, hydrochloric acid, zinc acetate, nickel acetate, sodium hydroxide, Na2EDTA, thioacetamide and 1-ethyl-3-methylimidazolium hexafluorophosphate ([EMIM][PF6]) were purchased from the Merck Company (Darmstadt, Germany).
2.2. Instrumentation
The chromatographic measurements were carried out with a KNAUER Smartline HPLC (Berlin, Germany) system equipped with LPG system, micro vacuum degasser, UV-Vis Detector (2550: was set at 220 nm) and a Zorbax SB-C18 column (150 × 4.0 mm, 5 μm: Agilent Technologies, USA). The chromatographic calculations were performed by using the EZChrom Elite system. Determination of carbofuran and propoxur were performed under the optimum separation conditions by HPLC with isocratic binary mobile phase consisting of water/acetonitrile (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) with flow rate of 1 mL min−1. An ultrasonic device (TECNO-GAZ, 60 Hz, 130 W, Parma, Italy) equipped with a digital timer and a temperature controller was used for the synthesis of the sorbent and desorption of the analytes. A digital pH meter (InoLab pH 730, Germany) was employed for the pH measurements.
:30, v/v) with flow rate of 1 mL min−1. An ultrasonic device (TECNO-GAZ, 60 Hz, 130 W, Parma, Italy) equipped with a digital timer and a temperature controller was used for the synthesis of the sorbent and desorption of the analytes. A digital pH meter (InoLab pH 730, Germany) was employed for the pH measurements.
2.3. Preparation of ZnS-AC-NPs
The synthesize method of ZnS-AC-NPs with some modification was done according to the given procedure in literature.31 Typically, ZnS NPs were synthesized by adding 5 mL of [Zn(CH3COO)2·2H2O] (1.0 mol L−1) into 10 mL of a 0.15 mol L−1 Na2EDTA solution (pH = 6) under stirring followed by the addition of 25 mL of Na2S·5H2O (0.5 mol L−1) solution. Then, the volume of this mixture was diluted up to approximately to 100 mL upon the addition of double distilled water while the pH was adjusted to 6.0. Finally, 25.0 g of functionalized AC was added to the resulting solution and was mixed at 70 °C for about 30 minutes.32 In this step, the solution turned milky white indicating the initial formation of ZnS nanoparticles on the AC. In order to complete the reaction, the mixture was maintained at 70 °C for 10 hours and the ZnS-AC-NPs were filtered, extensively washed with double distilled water and dried at 100 °C in an oven for 8 hours.
2.4. Coated stir bar with ZnS-AC-NPs
The proposed stir bar was prepared by inserting a long steel wire (15 mm) into a capillary glass (∼20 mm) sealing its ends with flame. Then, the glass bar was cleaned with water and CH2Cl2 in an ultrasonic bath for 10 min. In the next step, the stir bar was immersed in 1.0 mol L−1 NaOH solution for 24 h for exposing the silanol groups to the glass surface33 and it was cleaned with water and HCl solution (0.1 mol L−1) and finally was dried at 50 °C for 5 h. In order to adhering the ZnS-AC-NPs to the surface, the stir bar was first coated with a film of the TEOS solution. This solution was prepared by mixing 2 mL of TEOS with a mixture of acetic acid (1.0 mL 1.0 mol L−1) and ethanol (2.0 mL) in a glass vial. Then the resulting mixture was homogenized for 15 min while a clear sol solution was obtained. In the next step, the treated glass stir bar was immersed vertically in the sol solution and was held for 25 minutes to coat the glass stir bar with TEOS sol. Finally, the coated stir bar was placed at the room temperature for 10 hours and TEOS coating was formed.
The ZnS-AC coated stir bar was prepared according to the method used for coating the carbon nanotubes.34,35 Typical, 1.0 mL of TEOS and 1.0 mL of ethanol was mixed in a glass vial and stirred for 5 min. Then, 0.5 mL of water and 200 μL of concentrated hydrochloric acid were added and the resulting solution was stirred for 20 min to promote the hydrolysis and the condensation reactions. Finally, 50 mg of [EMIM][PF6] was added to it and the solution was mixed for 100 min before the sol was completely formed. Then, 40 mg of ZnS-AC was added to 2.0 mL of this sol and the mixture was sonicated for 25 min. In next step, the coated stir bar was immersed in this mixture and was held for 25 min for deposition of ZnS-AC was on its surface. Then the modified bar was taken out to be dried at 60 °C for 24 hours. Prior to the application, the [EMIM][PF6] was removed from the silica matrix of the modified stir bar by cleaning it ultrasonically in ethanol and water, respectively, for 15 min. The removal of [EMIM][PF6] from the silica matrix results in coating with more large surface area, porosity and excellent exchange kinetic.36,37
2.5. Sample collection and preparation
To evaluate the accuracy and applicability of the proposed method, the extraction and determination of carbofuran and propoxur in different water samples, i.e., tap, mineral, and river waters were performed. The tap and mineral water samples were taken from our laboratory and local supermarket in Yasouj, Iran, respectively. In addition, the river water samples were taken from different field areas of the Beshar River at Yasouj, Iran (August 2015). The collected water samples were filtered through a 0.45 μm micropore membrane and were maintained in glass containers, then stored at a temperature of 4 °C until their analysis time.
2.6. Stir bar sorptive extraction procedure
The 10.0 mL of the sample solution containing 0.05 μg of carbofuran and propoxur with natural pH and 3.5% NaCl was transferred into a 25 mL round-bottom glass flask. The modified stir bar was immersed in the solution and the magnetic stirrer was stirring at 500 rpm for 22 min. Then, the solution was decanted and the stir bar was taken out, cleaned with water and dried. The extracted carbofuran and propoxur were desorbed by placing the modified stir bar into a cylindrical glass vial containing 320 μL of methanol with sonical agitation for 8 min. The methanol was evaporated under mild nitrogen stream, the sediment phase was dissolved in 50 μL of methanol and 20 μL of it was injected into the HPLC-UV system for the quantification of the analytes.
2.7. Calculating of the extraction recovery
In order to find the optimized extraction condition, the extraction recovery (ER) and enrichment factor (EF) was used to evaluate the optimum condition. ER% was defined as the percentage of the extracted analyte in the final extract (nf) per total analyte in the aqueous phase (naq). The ER%, as the analytical response, was calculated according to the following equation:| |
 | (1) |
where Vf, Cf and Vaq, Caq are the volume and concentrations of the carbofuran and propoxur in the final extract and aqueous phases, respectively. Cf was calculated according to the calibration graph of the standard solutions of the analytes in methanol.
3. Results and discussion
3.1. The selection of extraction sorbent
In preliminary experiments, the performance of coated stir bar with ZnS-AC-NPs and AC for extraction of carbofuran and propoxur was investigated. The obtained results confirmed that in contrast with the stir bar coated with ZnS-NPs on AC, the performance of AC loaded stir bar was much lower (∼27.0–40.0%). This observation contributed to the fact that ZnS-NPs have two reactive cites for reaction with carbofuran and propoxur which are as follows: (1) the reactive site of S in ZnS can react with π electrons of carbofuran and propoxur and (2) Zn site in ZnS-NPs also tends to interact with the structures containing OH group.
Fig. 1 shows XRD patterns of ZnS nanoparticles prepared at 70 °C for 10 h. The standard XRD pattern for ZnS (Joint Committee for Powder Diffraction Standards, JCPDS card no. 05-0566) is given at the bottom of Fig. 1. The exists of three broad peaks in the diffractogram at around 28.56° (111), 47.93° (220) and 57.12° (311) confirm the cubic lattice structure of ZnS. According to Debye–Scherrer equation and the full width at half-maximum (FWHM) of (111) peak, the average size of the ZnS-NPs was estimated to be about 60 nm. The size of this nanoparticle31 also increases the surface area of the sorbent for the extraction of carbofuran and propoxur. For these reasons, the ZnS nanoparticles loaded on the AC was chosen as the sorbent for the modification of the surface of stir bar.
 |
| | Fig. 1 X-ray diffraction (XRD) pattern of the ZnS nanoparticles. | |
3.2. Optimization of the SBSE condition
3.2.1. Selection of desorption solvent. To chosen the best desorption solvent and suitable behavior for HPLC analysis; different solvents including acetone, methanol and ethanol were initially examined. The extraction recovery (ER) was found to be as follows: acetone (65 ± 7%), methanol (82 ± 6%), and ethanol (77 ± 5%). According to these results and since methanol is more convenient for HPLC, it was chosen as the best desorption solvent in the subsequent experiments. Then, the experimental factors affecting the ER were studied by the FFD and respond surface methodology (RSM) in order to find the best conditions for the SBSE.
3.2.2. Optimization of extraction factors. The most popular first-order design is the two-level full or fractional factorial in which each factor is experimentally studied at only two levels. Due to their relative low cost and simplicity, a fractional factorial design (FFD) is useful in the initial optimization approach.38 An experimental FFD was built in order to screen the main factors affecting the ER of carbofuran and propoxur. It is to be attending that FFD does not determine the exact quantity of each factor, but with relatively a few experiments, it can provide important information concerning each factor.39 In FFD, the experiment runs should be performed in a random manner consisting two central points in order to minimize the effect of the uncontrolled variables and to estimate the pure error.40,41 According to preliminary studies and experiments, six factors including pH of sample solution (X1), ionic strength (NaCl% (w/v)) (X2), extraction time (minutes) (X3), volume of desorption solvent (μL) (X4), desorption time (minutes) (X5) as well as the stirring speed (rpm) (X6) were considered and a FFD (included 2f−2 = 16 experiments, which f is the number of factors) was developed to explore the important factors. The factor names, their symbols and levels have been listed in Table 1.
Table 1 Factors, symbols and levels in 26−2 FFD design matrix
| Factors |
Levels |
| Low (−1) |
Center point (0) |
High (+1) |
| (X1) pH of sample solution |
2 |
5 |
8 |
| (X2) ionic strength (NaCl% (w/v)) |
0 |
3 |
6 |
| (X3) extraction time (min) |
5 |
15 |
25 |
| (X4) volume of desorption solvent (μL) |
100 |
250 |
400 |
| (X5) desorption time (min) |
5 |
10 |
15 |
| (X6) stirring speed (rpm) |
400 |
600 |
800 |
| Run |
X1 |
X2 |
X3 |
X4 |
X5 |
X6 |
ER% |
| 1 |
2.0 |
0.0 |
5.0 |
100.0 |
5.0 |
400 |
45.0 |
| 2 |
8.0 |
0.0 |
5.0 |
100.0 |
15.0 |
400 |
39.0 |
| 3 |
2.0 |
6.0 |
5.0 |
100.0 |
15.0 |
800 |
70.0 |
| 4 |
8.0 |
6.0 |
5.0 |
100.0 |
5.0 |
800 |
65.0 |
| 5 |
2.0 |
0.0 |
25.0 |
100.0 |
15.0 |
800 |
55.0 |
| 6 |
8.0 |
0.0 |
25.0 |
100.0 |
5.0 |
800 |
58.0 |
| 7 |
2.0 |
6.0 |
25.0 |
100.0 |
5.0 |
400 |
72.0 |
| 8 |
8.0 |
6.0 |
25.0 |
100.0 |
15.0 |
400 |
81.0 |
| 9 |
2.0 |
0.0 |
5.0 |
400.0 |
5.0 |
800 |
59.0 |
| 10 |
8.0 |
0.0 |
5.0 |
400.0 |
15.0 |
800 |
43.0 |
| 11 |
2.0 |
6.0 |
5.0 |
400.0 |
15.0 |
400 |
65.0 |
| 12 |
8.0 |
6.0 |
5.0 |
400.0 |
5.0 |
400 |
78.0 |
| 13 |
2.0 |
0.0 |
25.0 |
400.0 |
15.0 |
400 |
80.0 |
| 14 |
8.0 |
0.0 |
25.0 |
400.0 |
5.0 |
400 |
74.0 |
| 15 |
2.0 |
6.0 |
25.0 |
400.0 |
5.0 |
800 |
83.0 |
| 16 |
8.0 |
6.0 |
25.0 |
400.0 |
15.0 |
800 |
92.0 |
| 17(C) |
5.0 |
3.0 |
15.0 |
250.0 |
10.0 |
600 |
89.0 |
| 18(C) |
5.0 |
3.0 |
15.0 |
250.0 |
10.0 |
600 |
91.0 |
All the experimental designs and the subsequent regressional analysis were calculated using the STATISTICA software (USA: version 7.0). The quality of the polynomial model equation was judged statistically by the coefficient of the determination R2, while its statistical significance was determined by the F-test. An analysis of variance (ANOVA) was executed in the design to assess the significance of the model.
Each experimental run was performed in triplicate and the average of them was reported. The analysis of these results produced the standardized main effect Pareto charts (P = 0.95) depicted in Fig. 2. The bar length in this figure is proportional to the significance of each factor for ER of carbofuran and propoxur. As can be seen from Fig. 2, the ionic strength of the sample solution, extraction time and the volume of desorption solvent have the most significant effects on the ER, so, they were analyzed in further assessment through central composite design (CCD). The pH, desorption time and stirring speed had no significant effect on the ER%. Thus, they were not considered in the further optimization process.
 |
| | Fig. 2 Standardized main effect Pareto chart for the FFD of screening experiment. Vertical line in the chart defines 95% confidence level. | |
Consequently, the optimum set of the experimental conditions was determined using sophisticated second-order models (RSM) which employ more than two levels to fit a full quadratic polynomial model. The CCD is one of the most common designs used to fit the quadratic models.42 A CCD combines two-level factorial points with axial points and at least one point at the center of the experimental region.42 The total number of the needed design points (N), is predicted by the following equation:38
where
f is the number of factors (here
f = 3); 2
f, the number of factorial points; 2
f, the number of axial points and
N0, the number of center points (
N0 = 2). Therefore, 16 experimental runs were needed for the CCD and the detailed of them are shown in
Table 2. The main effects as well as their interactions and quadratic effects have been considered in this design.
Table 2 Design matrix for the 23 central composite designs
| Factors |
Levels |
Star point α = 1.682 |
| Low (−1) |
Central (0) |
High (+1) |
−α |
+α |
| (X1) ionic strength (NaCl% (w/v)) |
1.2 |
3.0 |
4.8 |
0.0 |
6.0 |
| (X2) extraction time (min) |
12 |
18 |
24 |
7.9 |
28.1 |
| (X3) volume of desorption solvent (μL) |
150 |
250 |
350 |
81.8 |
418.2 |
| Runs |
X1 |
X2 |
X3 |
ER% |
| 1 |
−1 |
−1 |
−1 |
40.3 |
| 2 |
−1 |
−1 |
1 |
57.3 |
| 3 |
−1 |
1 |
−1 |
65.3 |
| 4 |
−1 |
1 |
1 |
79.2 |
| 5 |
1 |
−1 |
−1 |
53.5 |
| 6 |
1 |
−1 |
1 |
81.2 |
| 7 |
1 |
1 |
−1 |
74.5 |
| 8 |
1 |
1 |
1 |
91.5 |
| 9 |
−1.682 |
0 |
0 |
57.6 |
| 10 |
1.682 |
0 |
0 |
65.3 |
| 11 |
0 |
−1.682 |
0 |
53.6 |
| 12 |
0 |
1.682 |
0 |
92.4 |
| 13 |
0 |
0 |
−1.682 |
44.6 |
| 14 |
0 |
0 |
1.682 |
87.6 |
| 15(C) |
0 |
0 |
0 |
84.5 |
| 16(C) |
0 |
0 |
0 |
83.9 |
The most important effects of the main and the interaction factor were assessed by ANOVA (Table 3). A p-value less than 0.05 in Table 3 indicates the statistical significance of an effect at 95% confidence level.40 The suitability of the proposed model was also evaluated by calculating the determination coefficient (R2) and testing it for the lack of fit (LOF). The LOF p-value of 0.09 indicates that this parameter is not significantly relative to the pure error. Data analysis of the obtained results gave the following polynomial equation for of ER of carbofuran and propoxur:
| | |
ER% = 84.18 + 6.65X1 + 10.50X2 + 10.83X3 − 6.13X12 − 3.92X22 − 6.35X32 − 1.95X1X2
| (3) |
where
X1,
X2 and
X3 are the main factors described earlier. The fitness quality of the polynomial model equation was expressed by the coefficient of determination (experimental
R2 = 0.985 and adjusted
R2 = 0.963). The amount of the deviation around the mean is determined by
R2 and the closeness of the values of experimental and adjusted
R2 indicate a good relationship between the experimental data and the fitted model. The successful solvation of this equation when using the desirability function (DF) makes it possible to improve the performance of the proposed method for the extraction of carbofuran and propoxur. The plot of predicted ER% and raw residuals
versus the observed ER% have been shown in
Fig. 3a and b, respectively. Close inspection of
Fig. 3a reveals that the predicted ER% values generally fall on a straight line. According to
Fig. 3b, it is clear that there is no significant pattern followed in the plot of the residual
versus the predicted ER%. In general, these plots implied that the errors are normally distributed.
Table 3 Analysis of variance (ANOVA) for central composite design
| Source of variation |
Sum of square |
Df |
Mean square |
F-Value |
P-Value |
| X1 |
598.870 |
1 |
598.870 |
3327.053 |
0.011036 |
| X2 |
1507.014 |
1 |
1507.014 |
8372.301 |
0.006957 |
| X3 |
1602.116 |
1 |
1602.116 |
8900.646 |
0.006748 |
| X12 |
338.615 |
1 |
338.615 |
1881.196 |
0.014675 |
| X22 |
142.676 |
1 |
142.676 |
792.643 |
0.022603 |
| X32 |
375.617 |
1 |
375.617 |
2086.762 |
0.013934 |
| X1X2 |
30.420 |
1 |
30.420 |
169.000 |
0.048875 |
| X1X3 |
23.805 |
1 |
23.805 |
132.250 |
0.055219 |
| X2X3 |
23.805 |
1 |
23.805 |
132.250 |
0.055219 |
| Lack of fit |
63.389 |
5 |
12.678 |
70.432 |
0.090208 |
| Pure error |
0.180 |
1 |
0.180 |
|
|
| Total SS |
4354.644 |
15 |
|
|
|
 |
| | Fig. 3 (a) Plot of predicted value vs. observed value for extraction of carbofuran and propoxur. (b) Plot of residuals versus predicted response for extraction of carbofuran and propoxur. | |
The search of the proper values for the pH, the extraction time and the volume of methanol that maximizes the response (ER%) of the factors is important. Fig. 4 shows the most relevant fitted response surfaces for the CCD and depicts the response surface plots of ER% versus the affecting factors. The curvatures of these plots indicate the effective interaction between the factors. As shown in Fig. 4a–c, the ER% slightly increases and reaches a plateau in the middle of each factor with respect to the opposite axial. Thus, the recoveries of carbofuran and propoxur increased in the regions where the NaCl%, extraction time and volume of desorption solvent values were set at 3–4, 20–25 min and 300–330 μL, respectively.
 |
| | Fig. 4 Response surfaces for the 23 central composite designs: (a) Extraction time (min) – NaCl (w/v%); (b) NaCl (w/v%) – desorption solvent (μL); (c) desorption solvent (μL) – extraction time (min). | |
In final step, the desirability function (DF) was used to discover the best conditions for the extraction of carbofuran and propoxur.32 Each experimental response (Ui) and predicted response (Ûi) was transformed in order to produce a function for each individual response (di) and finally to find out a global function D that should maximize the optimum value of the effective factors with regards to their interactions. The response (U) was renewed into a particular desirability function (dfi) in the range of 0.0 (undesirable or minimum response) to 1.0 (desirable or ideal response). Then, the individual desirability scores dis were combined using a geometrical mean, which was optimized to find the optimum set of input factors:
| |
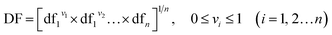 | (4) |
where df
i is the point to the desirability of each response
Ui (
i = 1, 2, 3, …,
n) and
vi represents the importance of responses. The individual DF of the predicted values for each dependent factor was computed
via the following equation where
α and
β are the lowest and highest obtained values of the response
i and
wi is the weight:
| |
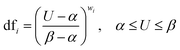 | (5) |
Fig. 5 summarizes the results of the DF for ER% of carbofuran and propoxur. The dashed lines and solid lines indicate the means and the standard deviation at 95% confidence limit for each factor, respectively. The highest values of ER% for each factor shows its optimum value. The profile for the predicted values and desirability option was used for the optimization process by specifying the DF for each dependent factor and assigning the predicted values over the range of 0.0 to 1.0.
 |
| | Fig. 5 Profiles for predicated values and desirability function for extraction recovery of carbofuran and propoxur. Dashed line indicated current values after optimization. | |
As shown in the Table 2, the maximum and minimum values of ER% for carbofuran and propoxur were 92.4 and 40.3%, respectively. The desirability values assigned for the DF of the dependent factors on ER% were 0.0 for minimum (40.3%), 0.5 for middle (66.3%) and 1.0 for maximum ER% (92.4%) which are depicted as three inflection points on the right hand side of Fig. 5. The individual desirability score of 1.0 selected as the target value for the optimization of the dependent factors is demonstrated at the bottom of the Fig. 5. In addition, the overall ER% of the carbofuran and propoxur plots with the observed level of factors is depicted at the top (left) of Fig. 5. These figures facilitate the search for the evaluation changes in the level of each factor simultaneously affecting the ER% and the overall desirability of the responses. According to these calculations as well as the target score of 1.0, the maximum value of ER% (95.4%) for carbofuran and propoxur was obtained at optimum values of NaCl of 3.5, extraction time of 22 min and desorption solvent of 320.0 μL of methanol. Finally, triplicate experiments were conducted using the optimized factors for validation while the ER% was found to be 94.8 ± 3.4%. The closeness of the experimental result to the data obtained from the desirability optimization analysis (94.4%) using CCD indicate that the combination of CCD and DF is suitable for simultaneous optimization of the extraction of carbofuran and propoxur.
3.3. Analytical performance of the SBSE-HPLC-UV
The quantitative parameters of the developed method such as linear range (LR), coefficient of correlation (r2), limit of detection (LOD), limit of quantification (LOQ), enrichment factors (EF) and precision were evaluated under optimum conditions as summarized in Table 4. Calibration curves were constructed by simultaneous extraction and determination of carbofuran and propoxur at six levels and a good linearity was obtained in the concentration range of 0.003–30 μg mL−1 and 0.002–30 μg mL−1 with the correlation coefficient of 0.998 and 0.999 for carbofuran and propoxur, respectively. The detection limit (LOD) of the method was calculated from 3σ, using the calibration curve data. The standard deviation of the blank (sb) was estimated from that of the residuals for this calculation. The LODs were 0.0004 and 0.0003 for carbofuran and propoxur, respectively. Precision (RSD%) of the method was determined by analyzing the standard samples containing 0.05 μg L−1 of carbofuran and propoxur (n = 6) and were found to be 3.5 and 4.8% for carbofuran and propoxur, respectively.
Table 4 Analytical performance of carbofuran and propoxur in water samples by the DLLME-HPLC method
| |
r2 |
LODa (μg mL−1) |
LOQb (μg mL−1) |
LRc (μg mL−1) |
EFd |
RSD (n = 6) |
| LOD: limit of detection. LOQ: limit of quantification. LR: linear range. EF: enrichment factor. |
| Carbofuran |
0.998 |
0.0004 |
0.0013 |
0.003–30 |
75 |
4.2 |
| Propoxur |
0.999 |
0.0003 |
0.0017 |
0.002–30 |
81 |
4.1 |
3.4. Analysis of real sample
The performance of the developed SBSE method was evaluated by the determination of carbofuran and propoxur in real water samples. After collection of the real samples, these samples were spiked at two levels and the accuracy of the method was determined through the recovery experiments that are shown in Table 5. The recoveries and relative standard deviations (RSDs) are in the range of 92.5–95.7%, indicating that the proposed method provides acceptable recoveries and precision for the determination of the carbofuran and propoxur in the real samples. The repeatability (intra-day) and reproducibility (inter-day) precision of the method were assessed according to the results of six replicates at level of 0.05 μg mL−1 of carbofuran and propoxur for six consecutive days. The values of intra-day relative standard deviation (RSD) and inter-day RSD were 5.5 and 7.3%, respectively. The chromatograms of the river and spiked river water samples at three concentration levels are shown in Fig. 6.
Table 5 Extraction recoveries of carbofuran and propoxur from water samples with the SBSE method (n = 3)
| Analyte |
Spiked (μg mL−1) |
Tap water |
River water |
Mineral water |
| Found (μg mL−1) |
ER ± RSD (%) |
Found (μg mL−1) |
ER ± RSD (%) |
Found (μg mL−1) |
ER ± RSD (%) |
| ND: not detect. |
| Carbofuran |
0.000 |
NDa |
— |
ND |
— |
ND |
— |
| 0.010 |
0.009 |
92.8 ± 3.8 |
0.009 |
92.5 ± 4.9 |
0.009 |
94.2 ± 3.6 |
| 0.100 |
0.095 |
95.4 ± 4.3 |
0.094 |
94.6 ± 3.8 |
0.095 |
95.7 ± 4.6 |
| Propoxur |
0.000 |
ND |
|
ND |
|
ND |
|
| 0.010 |
0.009 |
93.3 ± 3.8 |
0.009 |
93.7 ± 3.8 |
0.009 |
92.8 ± 4.1 |
| 0.100 |
0.094 |
94.5 ± 4.5 |
0.095 |
95.4 ± 4.3 |
0.093 |
93.8 ± 4.2 |
 |
| | Fig. 6 Chromatograms of extracted carbofuran (1) and propoxur (2) after SBSE-HPLC-UV at optimum extraction condition. (a) Tap water; (b), (c) and (d) tap water spiked with 0.02, 0.05 and 0.15 μg mL−1 of carbofuran and propoxur, respectively. | |
The comparison of SBSE procedures with different extraction techniques for the determination of target analytes is summarized in Table 6 [2, 4, 31]. The results showing that the developed method exhibits wide linear ranges, lower LODs as well as good reproducibility for the analytes.
Table 6 Comparison of the proposed method with other methods
| Method |
LR (μg mL−1) |
LOD (μg mL−1) |
RSD |
Ref. |
| DLLME-HPLC-UV |
0.005–10 |
0.0005 |
3.6 |
2 |
| MSA-DLLME-HPLC-DAD |
0.002–1 |
0.0004 |
4.5 |
4 |
| DNUM-HPLC-UV |
0.007–10 |
0.0020 |
4.5 |
31 |
| SBSE-HPLC-UV |
0.003–30 |
0.0003 |
4.2 |
This work |
4. Conclusion
This study demonstrates that the stir bar coated with ZnS-AC-NPs has a good capability for the extraction of carbofuran and propoxur. The prepared stir bar compared with the other methods is inexpensive, simple and can be used for the extraction of polar organic compounds. The combination of RSM and DF result in rapid and accurate optimization of simultaneous extraction of carbofuran and propoxur. In general, the developed SBSE method has provided an efficient microextraction technique with very low detection limit, good precision, and efficient recovery for the simultaneous separation and preconcentration of carbofuran and propoxur from water samples.
Acknowledgements
The authors express their appreciation to the Behbahan Faculty of Medical Sciences for financial support of this work.
References
- C. C. Wu, C. Chu, Y. S. Wang and H. S. Lur, J. Environ. Sci. Health, Part B, 2009, 44, 58–68 CrossRef CAS PubMed.
- S. Khodadoust and M. R. Hadjmohammadi, Anal. Chim. Acta, 2011, 699, 113–119 CrossRef CAS PubMed.
- S. Khodadoust, M. Ghaedi and M. R. Hadjmohammadi, Talanta, 2013, 116, 637–646 CrossRef CAS PubMed.
- X. Wang, J. Cheng, H. Zhou, X. Wang and M. Cheng, Anal. Chim. Acta, 2013, 787, 71–77 CrossRef CAS PubMed.
- S. Qian, Y. Leng and H. Lin, RSC Adv., 2016, 6, 7902–7907 RSC.
- J. Cheng, Y. T. Xia, Y. W. Zhou, F. Guo and G. Chen, Anal. Chim. Acta, 2011, 701, 86–91 CrossRef CAS PubMed.
- A. Santalad, L. Zhou, F. J. Shang, D. Fitzpatrick, R. Burakham, S. Srijaranai, J. D. Glennon and J. H. T. Luong, J. Chromatogr. A, 2010, 1217, 5288–5297 CrossRef CAS PubMed.
- H. Rashidi Nodeh, W. A. Wan Ibrahim, M. M. Sanagi and H. Y. Aboul-Enein, RSC Adv., 2016, 6, 24853–24864 RSC.
- A. A. Pebdani, A. M. Haji Shabani, S. Dadfarnia and S. Khodadoust, Spectrochim. Acta, Part A, 2015, 147, 26–30 CrossRef CAS PubMed.
- C. A. Harvey, J. C. Carter, J. R. Ertel, C. T. Alviso, S. C. Chinn and R. S. Maxwell, J. Chromatogr. A, 2015, 1401, 1–8 CrossRef CAS PubMed.
- Z. Zhong, G. Li, Z. Luo, Z. Liu, Y. Shao, W. He, J. Deng and X. Luo, Anal. Chim. Acta, 2015, 888, 75–84 CrossRef CAS PubMed.
- J. Chen, R. P. Liang, X. N. Wang and J. D. Qiu, J. Chromatogr. A, 2011, 1409, 268–276 CrossRef PubMed.
- E. Guerra, M. Celeiro, J. P. Lamas, M. Llompart and C. Garcia-Jares, J. Chromatogr. A, 2015, 1415, 27–37 CrossRef CAS PubMed.
- M. Liu, M. Li, B. Qiu, X. Chen and G. Chen, Anal. Chim. Acta, 2010, 663, 33–38 CrossRef CAS PubMed.
- E. Baltussen, P. Sandra, F. David and C. A. Cramers, J. Microcolumn Sep., 1999, 11, 737–747 CrossRef CAS.
- A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria and L. A. Fernández, J. Chromatogr. A, 2007, 1174, 40–49 CrossRef CAS PubMed.
- H. Yang, M. Xu, L.-X. Guo, H.-F. Ji, J.-Y. Wang, B.-P. Lin, X.-Q. Zhang and Y. Sun, RSC Adv., 2015, 5, 7304–7310 RSC.
- C. Hu, M. He, B. Chen and B. Hu, J. Chromatogr. A, 2013, 1275, 25–31 CrossRef CAS PubMed.
- X. J. Huang, J. B. Lin and D. X. Yuan, J. Chromatogr. A, 2009, 1216, 3508–3511 CrossRef CAS PubMed.
- M. Rezakazemi, A. Vatani and T. Mohammadi, RSC Adv., 2015, 5, 82460–82470 RSC.
- P. L. Kole, J. Millership and J. C. McElnay, Talanta, 2011, 85, 1948–1958 CrossRef CAS PubMed.
- B. Barfi, A. Asghari, M. Rajabi and N. Mirkhani, RSC Adv., 2015, 5, 106574–106588 RSC.
- J. G. Hou, Q. Ma, X. Z. Du, H. L. Deng and J. Z. Gao, Talanta, 2004, 62, 241–246 CrossRef CAS PubMed.
- C. Hu, M. He, B. Chen and B. Hu, J. Chromatogr. A, 2015, 1394, 36–45 CrossRef CAS PubMed.
- Y. Ma, Y. Zheng and J. P. Chen, J. Colloid Interface Sci., 2011, 354, 785–792 CrossRef CAS PubMed.
- A. Amiri Pebdani, S. Khodadoust, M. S. Talebianpoor, H. R. Zargar and V. Zarezade, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1008, 146–155 CrossRef PubMed.
- N. Gilart, R. M. Marce, F. Borrull and N. Fontanals, TrAC, Trends Anal. Chem., 2014, 54, 11–23 CrossRef CAS.
- A. Amiri Pebdani, S. Dadfarnia, A. M. Haji Shabani, S. Khodadoust and S. haghgoo, J. Chromatogr. A, 2016, 1437, 15–24 CrossRef PubMed.
- A. Kumar, Gaurav, A. K. Malik, D. K. Tewary and B. Singh, Anal. Chim. Acta, 2008, 610, 1–14 CrossRef CAS PubMed.
- S. Khodadoust and M. Ghaedi, Spectrochim. Acta, Part A, 2014, 133, 87–92 CrossRef CAS PubMed.
- S. Khodadoust, M. S. Talebianpoor and M. Ghaedi, J. Sep. Sci., 2014, 37, 3117–3124 CrossRef CAS PubMed.
- N. Zhang, L. Y. Wang, H. Liu and Q. K. Cai, Surf. Interface Anal., 2008, 40, 1190–1194 CrossRef CAS.
- B. Barfi, A. Asghari, M. Rajabi, S. Sabzalian, F. Khanalipoor and M. Behzad, RSC Adv., 2015, 5, 31930–31941 RSC.
- X. Y. Song, Y. P. Shi and J. Chen, Talanta, 2012, 100, 153–161 CrossRef CAS PubMed.
- Z. Es’haghi, Z. Rezaeifar, G. H. Rounaghi, Z. Alian Nezhadi and M. A. Golsefidi, Anal. Chim. Acta, 2011, 689, 122–128 CrossRef PubMed.
- N. Zhang and B. Hu, Anal. Chim. Acta, 2012, 723, 54–60 CrossRef CAS PubMed.
- M. Ebrahimi, Z. Es'haghi, F. Samadi and M. S. Hosseini, J. Chromatogr. A, 2011, 1218, 8313–8321 CrossRef CAS PubMed.
- M. Ghaedi, M. Roosta, S. Khodadoust and A. Daneshfar, J. Chromatogr. Sci., 2015, 53, 222–1231 Search PubMed.
- R. López, F. Goñi, A. Etxandia and E. Millán, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 846, 298–305 CrossRef PubMed.
- E. Morgan, Chemometrics: Experimental Design, Wiley, London, 1991 Search PubMed.
- G. E. P. Box and K. B. Wilson, J. Roy. Stat. Soc. B, 1951, 13, 1–45 Search PubMed.
- S. Khodadoust and M. Ghaedi, J. Sep. Sci., 2013, 36, 1734–1742 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 









