
Preparation and characterization of a covalent edge-functionalized lipoic acid–MoS2 conjugate†
Sang-Yong
Ju  *a
*a
*a
Abstract
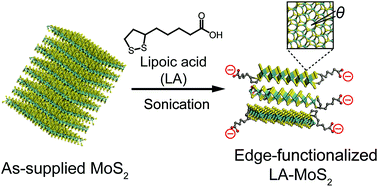
Covalent functionalization of semiconducting MoS2 is important in order to broaden the optoelectronic applications of this material. In the current study, we developed a one-step method for covalent edge functionalization of semiconducting MoS2 with lipoic acid (LA). The process, which utilizes a simple sonochemical aqueous dispersion approach, generates a LA–MoS2 conjugate that has a few layered nanoflake structure with a width of a few hundred nanometers and that display an intact crystalline basal plane. The MoS2 conjugate has the tendency to stack and fold with random relative angles owing to the presence of LA edge linked groups.
 Please wait while we load your content...
Please wait while we load your content...

